Volume 4, Number 2—June 1998
Dispatch
Dual Infection with Ehrlichia chaffeensis and a Spotted Fever Group Rickettsia: A Case Report
Figure
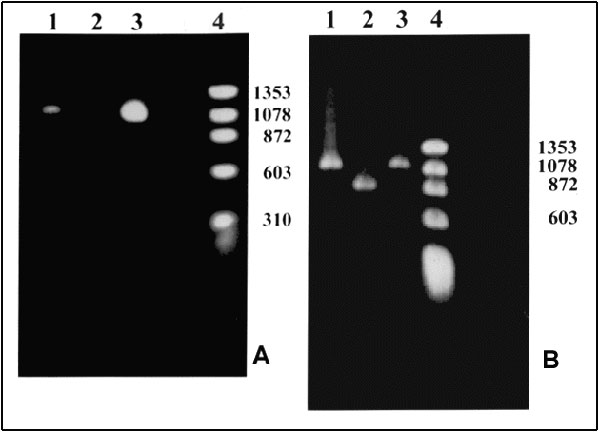
Figure. Polymerase chain reaction (PCR) products. A. With primers for the NadA gene. Lane 1: Patient's sample with an approximately 1.02-kb product; Lane 2: Negative control; Lane 3: Positive control. Ehrlichia chaffeensis (Arkansas strain)–infected DH82 cells; Lane 4: Molecular size markers: fX174 phage DNA cleaved with HaeIII. B. With nested primers for the 120 kDa protein gene. Lane 1: Patient's sample with an approximately 1.1-kb product; Lane 2: Positive control. E. chaffeensis (Sapulpa strain)–DH82 infected cells; Lane 3: Positive control. E. chaffeensis (Arkansas strain)–infected DH82 cells; Lane 4: Molecular size markers: fX174 phage DNA cleaved with HaeIII. Amplification conditions. Ten microliters of the patient's DNA was added to a 90-µl PCR reaction tube containing 10 µl of 10X PCR buffer (Boehringer Mannheim, Indianapolis, IN), 1 µl of PCR primers ECHNADA1 and pXCR6, final concentration 1 µM each; 2 µl deoxynucleotide triphosphates (final concentration, 200 µM); and 1 µl of Taq polymerase (Boehringer Mannheim, Indianapolis, IN). Water was added to bring the volume to 100 µl. The mixture was placed in a Progene FPROGO2Y thermocycler (Techne, Princeton, NJ). A DNA lysate prepared from E. chaffeensis-infected DH82 cells was used as a positive control. The cycling program consisted of 3 min at 94°C followed by 30 cycles, each of 30 sec at 94°C, 1 min at 55°C, and 2 min at 72°C, and an additional cycle with an extension step of 3 min at 72°C. To confirm the identity of the PCR product, we sequenced the 590 bp of the nadA gene product. After amplification, the PCR products were purified by QIAquick, a PCR purification kit (QIAgen, Santa Clarita, CA). The nucleotide sequence was then determined by the dideoxynucleotide method of cycle sequencing with Taq polymerase (ABI Prism 377 DNA sequencer, Perkin-Elmer Corp., Foster City, CA). The sequencing reaction was carried out for each strand of DNA. The sequencing data were analyzed by Genetics Computer Group, Wisconsin Package software; 99.8% identity of the 590-bp overlap with nadA nucleotide sequence of Arkansas strain (Gen Bank accession number U90900) was obtained (8).
References
- Nadelman RB, Horowitz HW, Hsieh T-C, Wu JM, Aguero-Rosenfeld ME, Schwartz I, Simultaneous human granulocytic ehrlichiosis and Lyme borreliosis. N Engl J Med. 1997;337:27–30. DOIPubMedGoogle Scholar
- Krause PJ, Telford SR III, Spielman A, Sikand V, Ryan R, Christianson D, Concurrent Lyme disease and babesiosis: evidence for increased severity and duration of illness. JAMA. 1996;275:1657–60. DOIPubMedGoogle Scholar
- Procop GW, Burchette JL, Howell DN, Sexton DJ. Immunoperoxidase and immunofluorescent staining of Rickettsia rickettsii in skin biopsy. A comparative study. Arch Pathol Lab Med. 1997;121:894–9.PubMedGoogle Scholar
- Philip RN, Casper EA, Ormsbee RA, Peacock MG, Burgdorfer W. Microimmunofluorescence test for the serological study of Rocky Mountain spotted fever and typhus. J Clin Microbiol. 1976;3:51–61.PubMedGoogle Scholar
- Dawson JE, Rikihisa Y, Ewing SA, Fishbein DB. Serologic diagnosis of human ehrlichiosis using two Ehrlichia canis isolates. J Infect Dis. 1991;163:564–7.PubMedGoogle Scholar
- Lange JV, Walker DH, Wester TB. Documented Rocky Mountain spotted fever in wintertime. JAMA. 1982;247:2403–4. DOIPubMedGoogle Scholar
- Iqbal Z, Rikihisa Y. Re-isolation of Ehrlichia canis from blood and tissue of dogs after doxycyline treatment. J Clin Microbiol. 1994;32:1644–9.PubMedGoogle Scholar
- Xu X-J, Walker DH. Sequence and characterization of an Ehrlichia chaffeensis gene encoding 314 amino acids highly homologous to the NAD A enzyme. FEMS Microbiol Lett. 1997;154:53–8. DOIPubMedGoogle Scholar
- Xu X-J, Crocquet-Valdes PA, Walker DH. Cloning and sequencing of the gene for 120-kDA immunodominant surface protein of Ehrlichia chaffeensis. Gene. 1997;184:149–54. DOIPubMedGoogle Scholar
- Xu X-J, Piesman JF, Olson JG, Walker DH. Geographic distribution of different genetic types of Ehrlichia chaffeensis. Am J Trop Med Hyg. 1997;56:679–80.PubMedGoogle Scholar
- Sexton DJ, Corey GR. Rocky Mountain spotless and almost spotless fever: a wolf in sheep's clothing. Clin Infect Dis. 1992;15:439–48.PubMedGoogle Scholar
- Anderson BE, Sims KG, Olson JG, Childs JE, Piesman JF, Happ CM, Amblyomma americanum: a potential vector of human ehrlichiosis. Am J Trop Med Hyg. 1993;49:239–44.PubMedGoogle Scholar
- Schubert JH. Serologic titers in rickettsial infection as affected by antibiotic treatment. Pub Health Lab. 1952;10:38–41.
- Davis JP, Wilfert CM, Sexton DJ, Burgdorfer W, Casper EA, Philip RN. Serologic comparison of R. rickettsii isolated from patients in Montana and North Carolina. In: Rickettsiae and rickettsial diseases. New York: Academic Press, Inc.; 1981 p. 139-47.
- Pancholi P, Kolbert CP, Mitchell PD, Reed KD Jr, Dumler JS, Bakken JS, Ixodes dammini as a potential vector of human granulocytic ehrlichiosis. J Infect Dis. 1995;172:1007–12.PubMedGoogle Scholar
- Telford SR III, Dawson JE, Katavolos P, Warner CK, Kolbert CP, Persing DH. Perpetuation of the agent of human granulocytic ehrlichiosis in a deer tick-rodent cycle. Proc Natl Acad Sci U S A. 1996;93:6209–14. DOIPubMedGoogle Scholar
- Schwartz I, Fish D, Daniels TJ. Prevalence of the rickettsial agent of human granulocytic ehrlichiosis in ticks from a hyperendemic focus of Lyme disease. N Engl J Med. 1997;336:49–50. DOIGoogle Scholar
- Ewing SA, Dawson JE, Kocan AA, Barker RW, Warner CK, Panciera RJ, Experimental transmission of Ehrlichia chaffeensis (Rickettsiales: Ehrlichieae) among white-tailed deer by Amblyomma americanum (Acarei: Ixodidae). J Med Entomol. 1995;32:368–74.PubMedGoogle Scholar
- Anderson BE, Sumner JW, Dawson JE, Tzianabos T, Greene CR, Olson JC, Detection of the etiologic agent of human ehrlichiosis by polymerase chain reaction. J Clin Microbiol. 1992;30:775–80.PubMedGoogle Scholar
- Dawson JE, Fishbein DB, Eng TR, Redus MA, Greene NR. Diagnosis of human ehrlichiosis with the indirect fluorescent antibody test: kinetics and specificity. J Infect Dis. 1990;162:91–5.PubMedGoogle Scholar