Volume 5, Number 1—February 1999
Research
Genetic Diversity and Distribution of Peromyscus-Borne Hantaviruses in North America
Cite This Article
Citation for Media
Abstract
The 1993 outbreak of hantavirus pulmonary syndrome (HPS) in the southwestern United States was associated with Sin Nombre virus, a rodent-borne hantavirus; The virus' primary reservoir is the deer mouse (Peromyscus maniculatus). Hantavirus-infected rodents were identified in various regions of North America. An extensive nucleotide sequence database of an 139 bp fragment amplified from virus M genomic segments was generated. Phylogenetic analysis confirmed that SNV-like hantaviruses are widely distributed in Peromyscus species rodents throughout North America. Classic SNV is the major cause of HPS in North America, but other Peromyscine-borne hantaviruses, e.g., New York and Monongahela viruses, are also associated with HPS cases. Although genetically diverse, SNV-like viruses have slowly coevolved with their rodent hosts. We show that the genetic relationships of hantaviruses in the Americas are complex, most likely as a result of the rapid radiation and speciation of New World sigmodontine rodents and occasional virus-host switching events.
Hantaviruses, rodent-borne RNA viruses, can be found worldwide. The Old World hantaviruses, such as Hantaan, Seoul, and Puumala, long known to be associated with human disease, cause hemorrhagic fever with renal syndrome of varying degrees of severity (1). After hantavirus pulmonary syndrome (HPS) was discovered in the southwestern United States in 1993 (2-4), intensive efforts were begun to detect and characterize hantaviruses in North America and determine their public health importance (5). As of January 1999, 205 HPS cases had been confirmed in 30 states of the United States, and 30 cases had been confirmed in three provinces of Canada; most cases occurred in the western regions of both countries. While Sin Nombre virus (SNV) has been identified as the cause of most HPS cases in North America, an increasingly complex array of additional hantaviruses has appeared (Table 1).
Surveys of rodents for hantavirus antibody have shown hantavirus-infected rodents in most areas of North America (3;6-9; Ksiazek et al., unpub. data; Artsob et al., unpub data). Serologic evidence of hantavirus infection has been found in North American rodents of the family Muridae. Most North American hantaviruses are associated with the subfamily Sigmodontinae; only a small number are associated with the subfamilies Arvicolinae or Murinae. To determine the number and distribution of hantaviruses in North America, we conducted a nucleotide sequence analysis of a polymerase chain reaction (PCR) fragment amplified from a large number of representative HPS patient and hantavirus-infected rodent samples from throughout the region. We focused on the North American viruses (particularly those associated with Peromyscus species rodents), although the nucleotide sequences of many hantaviruses from South America and elsewhere were included as outgroups to increase the resolution of the analysis.
Figure 1
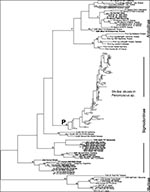
Figure 1. Overall hantavirus phylogenetic tree based on analysis of a 139 nucleotide fragment of the G2 coding region of the virus M segment. All newly reported sequences are shown bolded. The three...
Figure 2

The nucleotide sequences of 139 bp fragments of the G2 encoding region of virus M segments amplified by reverse transcriptase-PCR (RT-PCR) from 288 hantavirus-infected rodent and human samples were compiled from Genbank sources or from data reported here. Details of the specimen selection and methods of genomic analysis are provided in the Appendix. The Genbank Technical Appendix 1 of those sequences published earlier can be accessed here. The entire aligned Technical Appendix 2, including 130 newly presented sample sequences, is also available here. These sequences include those derived from 229 SNV-like viruses associated with Peromyscus species rodents from throughout North America. Maximum parsimony analysis of the aligned sequences was conducted with PAUP (12; Appendix), which resulted in a reasonably well-defined tree topology with several distinct lineages of SNV-like viruses and other clearly discernable hantaviruses (Figure 1, Figure 2). Bootstrap analysis showed that while several of the major nodes of the tree were not well supported (values of 50% or less), many others were robust (values of 70% or higher) (Figure 1, Figure 2). In most phylogenetic analyses, bootstrap values provide highly conservative estimates of the probability of correctly inferring the corresponding clades (13). Bootstrap values of 70% or higher corresponded to a probability of 95% or higher that the corresponding clade was correctly identified. Values of 50% or lower corresponded to a probability of 65% or lower that the clade was correctly identified (13).
As expected on the basis of earlier nucleotide sequence analysis of a limited number of complete S or M hantavirus genome segments or virus genome fragments (5), the evolutionary relationships among hantaviruses were closely correlated with those of their known or suspected primary rodent reservoirs (Figure 1; Table 1). Hantaviruses associated with subfamily Murinae rodents (Hantaan, Dobrava, Seoul, and Thailand viruses) are clearly separated from those associated with Arvicolinae and Sigmodontinae rodents. The Arvicolinae-associated viruses (Puumala, Khabarovsk, Tula, Isla Vista, Prospect Hill [PH], and PH-like viruses]) form a reasonably well-supported clade, but the phylogenetic position of this group relative to the Murinae- and Sigmodontinae-associated viruses is not well resolved.
The New World hantaviruses of the Arvicolinae group, primarily associated with Microtus species voles, include not only the classic PH virus (labeled PHV-1), originally isolated from M. pennsylvanicus in Maryland (14,15), and two other distinct PH-like virus lineages recently found in this vole species in North Dakota (R737 and R731; R742), but also Isla Vista virus in M. californicus, PH-like hantavirus lineages in M. ochrogaster in North Dakota (R812 and R789), and M. montanus in Wyoming and Nevada (3485; LY-R2312) (16,17). Virus phylogenetic placement is not clearly correlated with Microtus species of origin, indicating that either spill-over infection or host switching may occur with these viruses. An apparent example exists in the Ohio rodent samples of spill-over of a PH-like virus infection from Microtus species rodents to a deer mouse Peromyscus maniculatus (Pm1047). These viruses have not been associated with HPS cases.
The viruses associated with the subfamily Sigmodontinae rodents are highly diverse and are made up of several distinct viruses and lineages in North and South America. All viruses associated with Peromyscus species rodents form a well-supported distinct monophyletic clade (labeled P in Figure 1); these viruses constitute the major cause of HPS cases in North America. Other HPS-associated viruses in this group include Black Creek Canal virus, associated with Sigmodon hispidus. This virus, the cause of a single HPS case, has been genetically detected in cotton rats throughout southern Florida but, so far, nowhere else in the United States. Another genetically distinct virus, Muleshoe virus, has been identified in S. hispidus from the western part of its range (18), but sequences were not available for comparison at the time of our analysis. Caño Delgadito virus, found in S. alstoni in Venezuela (19), appears to be monophyletic with Black Creek Canal viruses. However, bootstrap support for this relationship is low (lower than 50%). Reasonable support is found for the clade containing both these Sigmodon sp.-associated viruses and the Bayou viruses, present in Oryzomys palustris throughout the southeastern United States from the Atlantic coast to Texas (20-22). Bayou viruses have been associated with three HPS cases (20-22). El Moro Canyon virus has been found in numerous harvest mice (Reithrodontomys megalotis) throughout the southwestern United States but has also been found in other rodents (e.g., WA-R2025, in M. montanus), presumably indicating spill-over infections (16,18,23,24). So far, these viruses have not been associated with human disease. The current phylogenetic analysis places these viruses in a distinct supported clade.
We analyzed hantaviruses that are also associated with HPS cases in South America and form a well-supported clade that encompasses viruses from Brazil, Argentina, and Paraguay, including the original Juquitiba virus detected in a human autopsy sample from an HPS patient in Brazil in 1993 (25-27). The rodent host for this virus is unknown. Two additional hantavirus lineages have been detected in more recent Brazilian HPS cases (Johnson and Nichol, unpub. data), suggesting that at least three genetically distinct hantaviruses are associated with HPS cases in Brazil. One of these lineages (b9618005) is phylogenetically closer to the Andes virus found in Argentina (28). Andes virus has recently been associated with several HPS cases in Patagonia; its likely host is Oligoryzomys longicaudatus (5,28,29). Finally, Laguna Negra viruses form a well-supported monophyletic lineage. This virus, associated with a large HPS outbreak in the Chaco region of Paraguay, is found in Calomys laucha rodents (10,30).
We analyzed 229 SNV-like viruses associated with Peromyscus species rodents; they form a well-supported (83%) clade (labeled P in Figure 1; details shown in Figure 2) and are distinct from other Sigmodontinae-associated hantaviruses. These SNV-like viruses include many classic SNVs, which are the major causes of HPS cases throughout the western and central United States and Canada, and are primarily associated with P. maniculatus. These viruses form a distinct, well-supported (78%) clade (labeled S in Figure 2), separate from other SNV-like viruses (Figure 2). Classic SNV 139 bp G2 fragments show up to an 18% nucleotide sequence divergence. Despite a number of exceptions, different genetic variants of SNV are grouped, generally speaking, by geography—an approximate geographic progression is apparent from the north and west toward the south and east, from the top of the tree down toward the node connecting these SNVs (labeled S in Figure 2). For instance, all samples from western Canada, including the Yukon, British Columbia, Alberta, Saskatchewan, and Manitoba are in the upper portion of this clade; two major lineages in California and Nevada (16,31) are also in this clade region. The lower part of the clade is dominated by viruses associated with the original Four Corners outbreak (New Mexico, Colorado, Utah, and Arizona) and other viruses from the Southwest, such as Kansas and Texas. Human HPS cases are represented throughout the SNV clade, indicating that these SNV variants can be associated with HPS illness.
In addition to recent samples, 30 SNV-like virus samples from the 1980s were included in the analysis to examine stability of the various SNV genetic lineages and their distribution (labeled H in Figure 2). Only small numbers of nucleotide differences, if any, were observed between old and recent virus sequences from the same geographic areas. The most striking example is the detection of identical viral G2 fragment sequences in rodents captured 12 years apart in New Mexico (Pm434) and Arizona (Pt AZ R29). Similarly, identical viral G2 sequences were found in rodents captured in eastern California in 1983 (our Pm435 and the previously published Sweetwater Canyon sequence [32]) and in human and rodent materials from eastern California and western Nevada sampled 10 or more years later (e.g., Humans CAH19 and NY-H575, and Pm LY-758, 786, and 792). Other examples include 1 of 139 and 2 of 139 nucleotide sequence differences between Washington rodent Pm432 (captured in 1980) and Pm206 and HPS case 0669 (sampled 16 years later), respectively; only 2 of 139 nucleotides are different between Pm428 from southern Oregon and Pm LY-R2302 from northern Nevada, despite capture 12 years apart. These and other data (6,7,32,33) suggest that SNV has been present in North America for a considerable time and has been relatively stably maintained in rodent populations.
The next most closely related viruses are those detected in the northeastern United States, referred to as New York virus (34). These viruses have been detected in two human HPS cases and in P. leucopus in New York and Rhode Island (Figure 2). The 139 nucleotide fragments of these viruses have up to 10.1% nucleotide variation, and they differ from classic SNVs by at least 11.5% at the nucleotide level. The next closest group contains viruses associated with several "forest form" subspecies of P. maniculatus throughout the eastern United States and Canada, including the cloudland deer mouse (P. maniculatus nubiterrae), which inhabits the Appalachian mountain region (35). These viruses can also be found in some P. leucopus in this region (e.g., rodent Pl 313 from Pennsylvania). Up to 17.3% nucleotide variation can be seen among the 139 nucleotide fragments of these viruses. The name Monongahela has been suggested for this virus lineage (36), which differs from New York and SN viruses by at least 8.6% and 10.8% nucleotide differences, respectively. Another distinct hantavirus lineage can be seen in P. maniculatus in Tennessee and has been associated with an HPS case (0027) in eastern North Carolina. These viruses are 7.9% different from one another at the nucleotide level for the 139 nucleotide fragment analyzed, and at least 12.2%, 14.4%, and 15.8% different from New York, Monongahela, and SN virus lineages, respectively. Additional distinct virus lineages, recently referred to as Blue River virus (37), can be detected in P. leucopus in Oklahoma (Pl 707), Indiana (e.g., Pl 9436372 and Pl 9436378), and Missouri (e.g., Pl 170). The Oklahoma lineage virus is 10.1%, 10.8%, 15.8% different from the viruses in the Missouri, Indiana, and Tennessee lineages.
In addition to identifying the distinct SNV-like viruses and virus genetic lineages throughout North America, our study provides data suggesting the likely site of infection and minimum incubation time for some HPS cases. As reported earlier (2), the HPS case labeled CO H5 was originally described as an Arizona case because the person was residing near Springerville, Arizona, when the illness began. However, the person had been living in Hesperus, Colorado, 11 days before disease onset. The PCR fragment amplified from the case autopsy specimen and from the P. maniculatus trapped at the household in Hesperus matched exactly and differed from those amplified from P. maniculatus in the Arizona location (Figure 2). Similarly, a patient (labeled human 0038) whose symptoms began in Los Angeles, California, had been in the Santa Fe, New Mexico, area 28 to 35 days before illness onset. Analysis of PCR fragments linked the source of infection to New Mexico, rather than to California (Figure 2).
The genetic data we present indicate a broad spectrum of genetic variants of SNV-like viruses throughout North America, associated primarily with Peromyscus rodents. Recent analysis of rodent mitochondrial DNA sequence differences suggests that the different SNV-like virus lineages are primarily associated with different Peromyscus species, and in some cases, with phylogenetically distinct subspecies or mitochondrial DNA haplotypes (Morzunov and Nichol, unpub. data; 37). For instance, the classic SNV and the Monongahela virus lineages are found associated with the "grassland form" and "forest form" of P. maniculatus, respectively (they represent different subspecies and appear phylogenetically distinct with respect to their mitochondrial DNA [Morzunov and Nichol, unpub. data]). The New York virus, and the Blue River virus lineages found in Indiana and Oklahoma, appear associated with genetically distinct P. leucopus populations (37). This pattern likely reflects microadaptation of the virus to the rodent host and not just geographic isolation of the virus variants. This view is supported by the observation that even in areas such as the eastern United States (particularly the Appalachian Mountain region), where P. maniculatus (forest form) and P. leucopus (eastern form) are sympatric and share microhabitat, extensive virus mixing between species is not seen; the Monongahela virus lineage is found predominantly in P. maniculatus, and the New York virus lineage in P. leucopus. Such data suggest that the broad correlation clearly evident between virus evolutionary relationships and those of their primary rodent reservoirs likely exists even at the finer level of closely related species and subspecies. However, the fact that the P. leucopus–associated New York virus appears phylogenetically closer to the P. maniculatus–associated viruses (SN and Monongahela) than to other P. leucopus–associated viruses (Blue River) suggests that this coevolutionary relationship is not absolute and that some species jumping (host-switching) may also have occurred. While the exact phylogenetic relationship of the SNV lineages to Monongahela, New York, and the other P. leucopus virus lineages is not well resolved by using the 139-bp G2 fragment we analyzed, analysis of more complete sequence data strongly supports a similar topology, placing New York virus firmly within the clade of P. maniculatus– borne viruses (37). This evidence, together with significant spill-over infection that sometimes occurs between sympatric rodents, illustrates the complexity of the hantavirus-host interactions.
This observation leads into another area of complexity, namely, the definition of distinct hantavirus serotypes or species. In the past, a newly identified arbovirus would be considered a distinct virus or virus serotype if a fourfold or greater two-way difference between this virus and previously recognized closely related viruses was obtained in virus neutralization assays. Despite the obvious biologic limitation (a single amino acid change can allow virus to escape from neutralization), this traditional criterion correlates remarkably well with more recent molecular data. One problem is that hantaviruses are generally difficult to isolate in tissue culture and are frequently noncytopathic, often making plaque assay analysis impractical (Table 1).
An attempt to define distinct virus species by using more widely used general criteria for the definition of biologic species is under way. Most defined species could be described as the lowest taxonomic unit that is geographically and ecologically contained, reproductively isolated, phylogenetically distinct, and self-sufficient. The apparent long-term maintenance and coevolution of phylogenetically distinct hantaviruses with different primary rodent reservoir species provides a foundation on which to build a hantavirus species definition. That is, if little host switching has occurred and if instead hantaviruses are associated with specific primary rodent reservoir species for many thousands of years, identification of a hantavirus in a unique primary rodent reservoir species would strongly suggest that in further analyses (e.g., nucleotide and amino acid sequence, cross-neutralization), it will be found to represent a new virus species. Hantaviruses maintained in rodent hosts from different genera (e.g., SNV in Peromyscus species rodents compared with Black Creek Canal virus in Sigmodon species rodents) will clearly meet the broad criteria for separate species status. This view is reinforced by recent data showing that stable reassortant viruses of different SNV genetic lineages can be readily detected in nature (31,38) and in tissue-culture mixed infections (39), but not in virus pairs such as SNV and Black Creek Canal virus (39). Difficulty can arise when trying to determine the species status of viruses maintained within rodent hosts of the same genera or species. So far, SN, New York, Monongahela, and Blue River viruses have been suggested as distinct hantaviruses with independent species names (5,36,37). The genetic analysis we present suggests that, as more hantavirus-infected Peromyscus species samples are analyzed, it is increasingly difficult to draw clear lines separating these virus species. The decision regarding whether to lump these viruses together as SNV-like viruses or to split them into separate species status will require the availability of neutralization data for several representatives of each virus, more detailed identification of the virus-host relationships, and more complete genetic characterization of both viruses and their hosts.
Rodent and HPS Case Materials
The newly described nucleotide sequences were derived from rodent materials collected as part of a nationwide survey of rodents for hantavirus antibodies (Ksiazek et al., unpub. data). Most of the human HPS-case blood and tissue autopsy samples were obtained and examined during the original investigation of an HPS outbreak in the Four Corners area of the southwestern United States in 1993 and as part of national surveillance for hantavirus disease throughout the United States from 1993 to 1997. Canadian rodent and HPS case materials were provided by the Laboratory Centre for Disease Control, Canada. Historic rodent samples were obtained from the Division of Biological Materials of the Museum of Southwestern Biology (Albuquerque, NM), University of New Mexico.
RNA Extraction, RT-PCR Amplification and Sequencing
Total RNA was extracted from human and rodent tissues, blood, or serum (2,10). Because of the hazardous nature of the virus, homogenization of rodent and human autopsy materials and extraction of RNA were performed in a certified class IIb laminar flow biosafety hood in Biosafety Level 3 containment. RNA was extracted from tissue or blood products by using acid guanidinium thiocyanate and phenol-chloroform and purified by using the RNaid Kit (Bio 101, La Jolla, CA). Nested RT-PCR assays were used to amplify DNA products containing a small fragment of the G2 coding region of M segment (2,10). Rodent and human samples were amplified separately, and all manipulations that might result in possible cross-contamination of samples were avoided. PCR products of correct size were sequenced with the same primers used for second-round PCR amplification in conjunction with various generations of sequencing kits available from Applied Biosystems, Inc. (Perkin Elmer, Foster City, CA). Sequences 139 nucleotides in length determined from each PCR product were used in phylogenetic analysis.
Oligonucleotide Primer Design
Oligonucleotide primers were used to generate DNA fragments from the G2 region of hantavirus M RNA (Table 2). In the initial phase of this project, amplification of hantavirus sequences from autopsy tissues of fatal HPS cases and hantavirus antibody-positive rodents in the southwestern United States used primers designed on the basis of nucleotide sequences conserved among PH and Puumala viruses (2). On the basis of SNV nucleotide sequences derived from these materials, new primers were designed and optimized for detection of SNV-like viruses associated with P. maniculatus (11). As more sequence data became available, additional generations of primers were refined that would detect hantaviruses from other geographic regions of the United States. The development of broadly reactive primers designed to detect hantaviruses associated with subfamily Sigmodontinae rodents (10) has eliminated the effort of amplifying RNA samples with many sets of primers.
Nucleotide Sequence and Phylogenetic Analyses
Compilation, alignment, and comparative nucleotide sequence analysis was carried out by using the Wisconsin Sequence Analysis Package, version 8.1 (Genetics Computer Group, Inc., Madison, WI) on a DEC 3000-500X AXP workstation (Digital Equipment Corp., Maynard, MA). Phylogenetic analysis was performed by maximum parsimony analysis using PAUP version 4.0 d52 (12) on a Power PC 9500. The size and complexity of the Technical Appendix 2 prevented the use of branch and bound search method and weighting of the data matrix based on transition:transversion bias. Maximum parsimony analysis of the hantavirus G2 fragment nucleotide sequences was carried out by using the heuristic search option. The initial unweighted analysis showed considerable homoplasy within the Technical Appendix 2. A successive approximations method was used in which characters were weighted by using the maximum value of their rescaled consistency index (12), and the heuristic search repeated. Bootstrap analysis was carried out by 500 replicates of the heuristic search with random resampling of the data. The analysis required several months of computer time; thus, it was not possible to include some recently published additional hantaviruses sequences. The nucleotide sequence Technical Appendix 2 we used is available in NEXUS format (compatible with most phylogenetic analysis software packages). A brief description of all samples analyzed is available in EXCEL 2.1 spreadsheet format (Technical Appendix 1). The phylogenetic tree of Figure 1 and Figure 2 can also be found at the same location on the website.
Ms. Monroe is a biologist with the Special Pathogens Branch, Division of Viral and Rickettsial Diseases, CDC. Her research focuses on molecular phylogenetics of hantaviruses and reverse genetic studies of Ebola virus.
Acknowledgments
We thank Ali Khan, Jim Mills, Jamie Childs, John Krebs, Tom Mather, Joe Camp, Fred Jannett, and many others for their efforts in collecting hantavirus infected materials. We also thank Kent Wagoner and Laura Morgan for assistance with graphics and database manipulations.
This work was supported in part by National Institutes of Health grants 5RO1AI36418-04 and 1PO1AI39808-01 through the University of Nevada-Reno.
References
- McKee KT Jr, LeDuc JW, Peters CJ. Hantaviruses. In: Belshe RB, editor. Textbook of human virology. 2nd ed. St. Louis (MO): Mosby; 1991. p. 615-32.
- Nichol ST, Spiropoulou CF, Morzunov SP, Rollin PE, Ksiazek TG, Feldmann H, Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science. 1993;262:914–7. DOIPubMedGoogle Scholar
- Childs JE, Ksiazek TG, Spiropoulou CF, Krebs JW, Morzunov S, Maupin GO, Serologic and genetic identification of Peromyscus maniculatus as the primary rodent reservoir for a new hantavirus in the southwestern United States. J Infect Dis. 1994;169:127–80.PubMedGoogle Scholar
- Duchin JS, Koster FT, Peters CJ, Simpson GL, Tempest B, Zaki SR, Hantavirus pulmonary syndrome: a clinical description of 17 patients with a newly recognized disease. N Engl J Med. 1994;330:949–55. DOIPubMedGoogle Scholar
- Schmaljohn C, Hjelle B. Hantaviruses: a global disease problem. Emerg Infect Dis. 1997;3:95–104. DOIPubMedGoogle Scholar
- Tsai TF, Bauer SP, Sasso DR, Whitfield SG, McCormick JB, Caraway CT, Serological and virological evidence of a Hantaan virus-related enzootic in the United States. J Infect Dis. 1985;152:126–36.PubMedGoogle Scholar
- Yanagihara R, Daum CA, Lee P-W, Baek L-J, Amyx HL, Gajdusek DC, Serological survey of Prospect Hill virus infection in indigenous wild rodents in the USA. Trans R Soc Trop Med Hyg. 1987;81:42–5. DOIPubMedGoogle Scholar
- Mills JN, Johnson JM, Ksiazek TG, Ellis BA, Rollin PE, Yates TL, Patterns of association with host and habitat: Antibody reactive with Sin Nombre virus in small mammals in the major biotic communities of the southwestern United States. Am J Trop Med Hyg. 1997;56:273–84.PubMedGoogle Scholar
- Mills JN, Johnson JM, Ksiazek TG, Ellis BA, Rollin PE, Yates TL, A survey of hantavirus antibody in small-mammal populations in selected United States national parks. Am J Trop Med Hyg. 1998;58:525–32.PubMedGoogle Scholar
- Johnson AM, Bowen MD, Ksiazek TG, Williams RJ, Bryan RT, Mills JN, Laguna Negra virus associated with HPS in western Paraguay and Bolivia. Virology. 1997;238:115–27. DOIPubMedGoogle Scholar
- Spiropoulou CF, Morzunov S, Feldmann H, Sanchez A, Peters CJ, Nichol ST. Genome structure and variability of a virus causing hantavirus pulmonary syndrome. Virology. 1994;200:715–23. DOIPubMedGoogle Scholar
- Swofford DL. PAUP*: phylogenetic analysis using parsimony (*and other methods) [computer program]. Version 4.0. Sinauer, Sunderland, MA; 1998.
- Hillis DM, Bull JJ. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol. 1993;42:182–92.
- Lee P, Amyx HL, Yanagihata R, Gajdusek DC, Goldgaber D, Gibbs CJ Jr. Partial characterization of Prospect Hill virus isolated from meadow voles in the United States. J Infect Dis. 1985;152:826–9.PubMedGoogle Scholar
- Parrington MA, Lee PW, Kang CY. Molecular characterization of the Prospect Hill virus M RNA segment: comparison with the M RNA segments of other hantaviruses. J Gen Virol. 1991;72:1845–54. DOIPubMedGoogle Scholar
- Rowe JE, St Jeor SC, Riolo J, Otteson EW, Monroe MC, Ksiazek TG, Coexistence of several novel hantaviruses in rodents indigenous to North America. Virology. 1995;213:122–30. DOIPubMedGoogle Scholar
- Song W, Torrez-Martinez N, Irwin W, Harrison FJ, Davis R, Ascher M, Isla Vista virus: a genetically novel hantavirus of the California vole Microtus californicus. J Gen Virol. 1995;76:3195–9. DOIPubMedGoogle Scholar
- Rawlings JA, Torrez-Martinez N, Neill SU, Moore GM, Hicks BN, Pichuantes S, Cocirculation of multiple hantaviruses in Texas, with characterization of the small (S) genome of a previously undescribed virus of cotton rats (Sigmodon hispidus). Am J Trop Med Hyg. 1996;55:672–9.PubMedGoogle Scholar
- Fulhorst CF, Monroe MC, Salas RA, Duno G, Utrera A, Ksiazek TG, Isolation, characterization, and geographic distribution of Caño Delgadito virus, a newly discovered South American hantavirus (family Bunyaviridae). Virus Res. 1997;51:159–71. DOIPubMedGoogle Scholar
- Morzunov SP, Feldmann H, Spiropoulou CF, Semenova VA, Rollin PE, Ksiazek TG, A newly recognized virus associated with a fatal case of hantavirus pulmonary syndrome in Louisiana. J Virol. 1995;69:1980–3.PubMedGoogle Scholar
- Ksiazek TG, Nichol ST, Mills JN, Groves MG, Wozniak A, McAdams S, Isolation, genetic diversity and geographic distribution of Bayou virus. Am J Trop Med Hyg. 1997;57:445–8.PubMedGoogle Scholar
- Hjelle B, Goade D, Torrez-Martinez N, Lang-Williams M, Kim J, Harris RL, Hantavirus pulmonary syndrome, renal insufficiency and myositis associated with infection by Bayou hantavirus. Clin Infect Dis. 1996;23:495–500.PubMedGoogle Scholar
- Hjelle B, Chavez-Giles F, Torrez-Martinez N, Yates T, Sarisky J, Webb J, Genetic identification of a novel hantavirus of the harvest mouse Reithrodontomys megalotis. J Virol. 1994;68:6751–4.PubMedGoogle Scholar
- Torrez-Martinez N, Song W, Hjelle B. Nucleotide sequence analysis of the M genomic segment of El Moro Canyon hantavirus: antigenic distinction from Four Corners hantavirus. Virology. 1995;211:336–8. DOIPubMedGoogle Scholar
- Nichol ST, Ksiazek TG, Rollin PE, Peters CJ. Hantavirus pulmonary syndrome and newly described hantaviruses in the United States. In: Elliott RM, editor. The Bunyaviridae. New York: Plenum Press; 1996. p. 269-80.
- da Silva MV, Vasconcelos MJ, Hidalgo NTR, Veiga APR, Canzian M, Marotto PCF, Rev Inst Med Trop Sao Paulo. 1997;39:231–4.PubMedGoogle Scholar
- Vasconcelos MJ, Lima VP, Iversson LB, Rosa MDB, Travassos Da Rosa APA, Travassos Da Rosa ES, Pulmonary syndrome in the rural area of Juquitiba, São Paulo metropolitan area, Brazil. Rev Inst Med Trop Sao Paulo. 1997;39:237–8.PubMedGoogle Scholar
- Lopez N, Padula P, Rossi C, Lazaro ME, Franze-Fernandez MT. Genetic identification of a new hantavirus causing severe pulmonary syndrome in Argentina. Virology. 1996;220:223–6. DOIPubMedGoogle Scholar
- Levis S, Rowe JE, Morzunov S, Enria DA, St Jeor S. New hantavirus causing hantavirus pulmonary syndrome in central Argentina. Lancet. 1997;349:998–9. DOIPubMedGoogle Scholar
- Williams RJ, Bryan RT, Mills JN, Palma RE, Vera I, de Velasquez F, An outbreak of hantavirus pulmonary syndrome in western Paraguay. Am J Trop Med Hyg. 1997;57:274–82.PubMedGoogle Scholar
- Henderson WW, Monroe MC, St Jeor SC, Thayer WP, Rowe JE, Peters CJ, Naturally occurring Sin Nombre virus genetic reassortants. Virology. 1995;213:602–10. DOIGoogle Scholar
- Nerurkar VR, Song JW, Song KJ, Nagle JW, Hjelle B, Jenison S, Genetic evidence for a hantavirus enzootic in deer mice (Peromyscus maniculatus) captured a decade before the recognition of hantavirus pulmonary syndrome. Virology. 1994;204:563–8. DOIPubMedGoogle Scholar
- Zaki SR, Khan AS, Goodman RA, Armstrong LR, Greer PW, Coffield LM, Retrospective diagnosis of hantavirus pulmonary syndrome, 1978-1993—Implications for emerging infectious diseases. Arch Pathol Lab Med. 1996;120:134–9.PubMedGoogle Scholar
- Hjelle B, Lee SW, Song W, Torrez-Martinez N, Song JW, Yanagihara R, Molecular linkage of hantavirus pulmonary syndrome to the white-footed mouse, Peromyscus leucopus: genetic characterization of the M genome of New York virus. J Virol. 1995;69:8137–41.PubMedGoogle Scholar
- Hall ER. Mammals of North America. New York: John Wiley and Sons; 1981.
- Song JW, Baek LJ, Nagle JW, Schlitter D, Yanagihara R. Genetic and phylogenetic analyses of hantaviral sequences amplified from archival tissues of deer mice (Peromyscus maniculatus nubiterrae) captured in the eastern United States. Arch Virol. 1996;141:959–67. DOIPubMedGoogle Scholar
- Morzunov SP, Rowe JE, Ksiazek TG, Peters CJ, St Jeor SC, Nichol ST. Genetic analysis of the diversity and origin of hantaviruses in Peromyscus leucopus mice in North America. J Virol. 1998;72:57–64.PubMedGoogle Scholar
- Schmaljohn AL, Li D, Negley DL, Bressler DS, Turell MJ, Korch GW, Isolation and initial characterization of a new found hantavirus from California. Virology. 1995;206:963–72. DOIPubMedGoogle Scholar
- Rodriguez LL, Owens JH, Peters CJ, Nichol ST. Genetic reassortment among viruses causing hantavirus pulmonary syndrome. Virology. 1998;242:99–106. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 5, Number 1—February 1999
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Stuart Nichol, Centers for Disease Control and Prevention, Special Pathogens Branch, Mail Stop G14, 1600 Clifton Rd., N.E., Atlanta, Georgia 30333, USA; fax: 404-639-1118
Top