Volume 10, Number 12—December 2004
Research
Venezuelan Equine Encephalitis Virus, Southern Mexico
José G. Estrada-Franco*, Roberto Navarro-Lopez†, Jerome E. Freier‡, Dionicio Cordova§, Tamara Clements¶, Abelardo Moncayo*, Wenli Kang*, Carlos Gomez-Hernandez#, Gabriela Rodriguez-Dominguez#, George V. Ludwig¶, and Nikos Vasilakis*
Figure 4
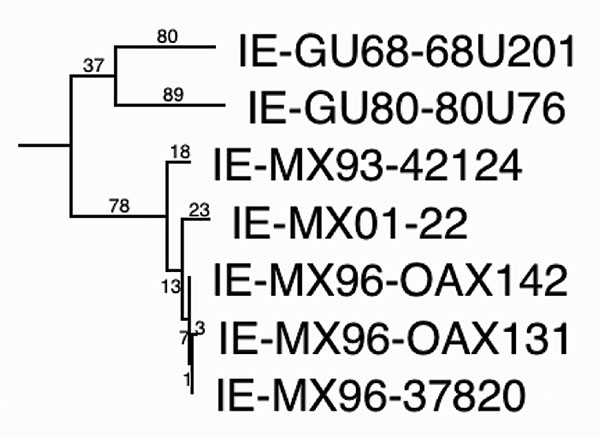
Figure 4. Maximum parsimony phylogenetic tree derived from complete genomic sequences showing relationships of the newly isolated Venezuelan equine encephalitis virus (VEEV) strain (MX01-22) to other strains from Mexico and Guatemala. Numbers indicate nucleotide substitutions assigned to each branch. All nodes had bootstrap values of 100%, except the OAX131-37820 (62%) and GU68-GU80 (<50%) groupings. Relative rates tests applied to the branches indicated a rate of nucleotide substitution in Mexico of 2.0–2.9 x 10–4 subst/nt/y since 1993, and 6.8 x 10–5 for the Guatemalan lineage from 1968–1980. These data suggest continuous circulation of VEEV in Mexico since 1993.
Page created: April 14, 2011
Page updated: April 14, 2011
Page reviewed: April 14, 2011
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.