Volume 10, Number 12—December 2004
Dispatch
Genome Sequence and Attenuating Mutations in West Nile Virus Isolate from Mexico
David W.C. Beasley* , C. Todd Davis*, Jose Estrada-Franco*, Roberto Navarro-Lopez†, Arturo Campomanes-Cortes†, Robert B. Tesh*, Nikos Vasilakis*, and Alan D.T. Barrett*
, C. Todd Davis*, Jose Estrada-Franco*, Roberto Navarro-Lopez†, Arturo Campomanes-Cortes†, Robert B. Tesh*, Nikos Vasilakis*, and Alan D.T. Barrett*
Figure 2
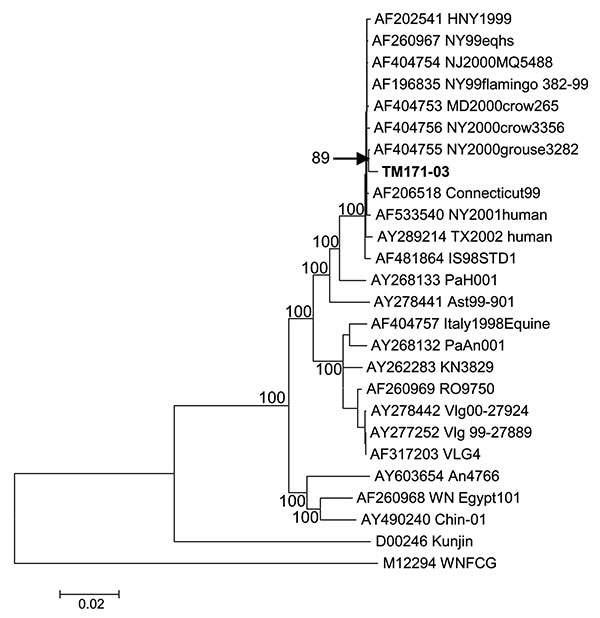
Figure 2. Neighbor-joining phylogenetic tree based on complete genome sequences of West Nile virus strains. Strain TM171-03 is indicated in bold text. The topology of maximum parsimony and maximum likelihood trees was essentially identical. Bayesian analysis also confirmed the close relationship between TM171-03 and NY00-grouse3282 sequences (data not shown). Bootstrap values are shown for major branches (500 replicates). GenBank accession numbers for sequences used to construct the tree are indicated on the branches.
Page created: April 14, 2011
Page updated: April 14, 2011
Page reviewed: April 14, 2011
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.