Volume 10, Number 9—September 2004
Research
Genotyping, Orientalis-like Yersinia pestis, and Plague Pandemics
Abstract
Three pandemics have been attributed to plague in the last 1,500 years. Yersinia pestis caused the third, and its DNA was found in human remains from the second. The Antiqua biovar of Y. pestis may have caused the first pandemic; the other two biovars, Medievalis and Orientalis, may have caused the second and third pandemics, respectively. To test this hypothesis, we designed an original genotyping system based on intergenic spacer sequencing called multiple spacer typing (MST). We found that MST differentiated every biovar in a collection of 36 Y. pestis isolates representative of the three biovars. When MST was applied to dental pulp collected from remains of eight persons who likely died in the first and second pandemics, this system identified original sequences that matched those of Y. pestis Orientalis. These data indicate that Y. pestis caused cases of Justinian plague. The two historical plague pandemics were likely caused by Orientalis-like strains.
Yersinia pestis, a group A bioterrorism agent (1), causes plague, a reemerging zoonotic disease transmitted to humans through flea bites and typically characterized by the appearance of a tender and swollen lymph node, the bubo (2). This organism has been subdivided into three biovars on the basis of their abilities to ferment glycerol and to reduce nitrate. Based on their current geographic niche and on historical records that indicate the geographic origin of the pandemics, researchers have postulated that each biovar caused a specific pandemic (2,3). Biovar Antiqua, from East Africa, may have descended from bacteria that caused the first pandemic, whereas Medievalis, from central Asia, may have descended from the bacteria that caused the second pandemic. Bacteria linked to the third pandemic are all of the Orientalis biovar (3). In this study, we tested this hypothesis for the first time by detecting biovars in ancient human remains.
No molecular biology–based method proved reliable and convenient for Y. pestis genotyping. Genome sequences of Y. pestis strain CO92, a Orientalis biovar, and Y. pestis strain KIM, a Medievalis biovar, are now available (4,5), which provides an opportunity to examine them for differences associated with the biovar and for genotyping. Genome analysis of the closely related Rickettsia prowazekii (6) and R. conorii (7) showed that intergenic spacers, which have been submitted to less evolutionary pressure than coding sequences, may be variable enough to differentiate closely related microorganisms. We, therefore, hypothesized that sequencing of several intergenic spacers would allow determination of a biovar-specific spacer pattern in Y. pestis. We named this method multiple spacer typing (MST). We first demonstrated that MST allowed biovar genotyping of a large collection of Y. pestis isolates and further applied it to the dental pulp collected from persons whose deaths are attributed to the first and second pandemics.
Bacterial Strains
Thirty-five strains representative of the three Y. pestis biovars (11 Antiqua isolates, 12 Medievalis isolates, and 12 Orientalis isolates) isolated from 1947 to 1996 from various host species in 13 countries are presented in Table 1. Nineteen of these isolates have been previously characterized by Achtman et al. (8). Nucleic acid was extracted as previously described (9), and species identification was confirmed for all the strains by partial sequencing of the rpob gene (10).
Spacer Sequence Database and Phylogenetic Analyses
We analyzed the complete genome sequences of Y. pestis strain CO92, biovar Orientalis (GenBank accession no. NC-003143) (4) and Y. pestis strain KIM, biovar Medievalis (GenBank accession no. NC-004088) (5), which were obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (11). We used the Primer3 program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) to determine the primer sequences specific for the genomic segments of interest (12). The primers flanked intergenic sequences of Y. pestis CO92 that exhibited large sequence differences with the homologous Y. pestis KIM strain sequences. We generated a list of Y. pestis CO92 intergenic sequences of 50 to 300 bp and carried out BLASTN searches to identify the homologous intergenic sequences in Y. pestis KIM strain by using the Y. pestis CO92 genes flanking the intergenic sequences as queries (13). When both genes flanking the intergenic sequence exhibited best-matches with the BLAST score >120 bits, we estimated the length of the corresponding intergenic sequence in the Y. pestis KIM strain. We then aligned the homologous intergenic sequences (<300 bp) and selected eight pairs of sequences with insertion or deletion divergence between the two Y. pestis biovars to locate the primers (Table 2). Two microliters of DNA extracted as previously described (9) were amplified in a 50-µL mixture containing 10 pmol of each primer; 200 mmol/L (each) dATP, dCTP, dGTP, dTTP (Invitrogen, Cergy-Pontoise, France); 1.5 U Taq DNA polymerase (Invitrogen); and 2.5 µL of a 50-mM solution of MgCl2 in 1 x Taq buffer. Each polymerase chain reaction (PCR) was performed in a T3 thermocycler (Biométra, Archamps, France) under the following conditions: an initial 5 min of denaturation at 95°C was followed by 39 cycles of denaturation for 30 s at 94°C, annealing for 30 s at 60°C, and extension for 1 min at 72°C. The amplification was completed by holding the reaction mixture for 5 min at 72°C to allow complete extension of PCR products. These products were purified by using the Multiscreen PCR plate (Millipore Corp., Bedford, MA), as described by the manufacturer. Sequencing reactions were carried out with a DNA sequencing kit (Big Dye Terminator Cycle Sequencing V2.0; PE Biosystem, Courtaboeuf, France), as described by the manufacturer. Sequencing products were purified and underwent electrophoresis with the 3100 Genetic Analyzer (Applied Biosystems) and aligned by using the multisequence alignment CLUSTALX version 1.8 (13).
For phylogenetic analyses, DNA sequences were aligned by using the CLUSTALW software, version 1.81 (13). Because variations among spacer sequences were only due to deletions (see below), every deletion was considered a unique molecular event that resulted in a unique mismatch, regardless of its length. For any position in the spacer sequence, identity of nucleotide was coded "1"; a mismatch was coded "0"; and a pairwise binary matrix was constructed with the MEGA 2.1 software package (14). Distance matrices were determined by using p-distance analysis and were used to infer dendrograms by the unweighted pair group method with arithmetic mean (UPGMA), maximum parsimony, and neighbor-joining, available in the MEGA 2.1 software package (14). We also used the maximum likelihood method within the PHYLIP software package (15).
Sources of Ancient DNA
Appendix Figure 1

Appendix Figure 1. Partial view of the grave in Dreux investigated in this work, which illustrates anthropologic features of a mass grave suitable for paleomicrobiology research.
The Anthropology Laboratory of Bordeaux University 1 studied a first series of 60 skeletons discovered during excavations in 1989 in Sens, France. The skeletons were buried in four adjoining mass graves, dated by radiocarbon to be from the 5th to 6th century A.D (16). The second series came from a cemetery in Dreux, France, where nine mass graves were identified during excavations in 1990 (Appendix Figure 1). Each grave contained 2–22 skeletons. Ceramic fragments, found in the burial site, dated the graves from the 12th to the 14th century A.D (17). Archeological data (burial patterns) and anthropologic data (absence of bone fractures, indications of sex and age of persons) supported the hypothesis that the two grave sites contained remains of persons who died during an epidemic. No historical written records for the Sens and Dreux grave sites exist, but comparisons with demographic models suggest that the graves resulted from the Justinian plague (6th–8th century) and the Black Death, respectively. The Saint-Côme and Saint-Damien site in Montpellier had been used as a church cemetery outside the city walls during the 9th to 17th centuries A.D (18). Four graves have been dated as having been dug in the 13th and late 14th centuries, on the basis of their position on top of a 13th-century remblai (a small hill created by burying bodies) and the fact that they were behind a wall that dated from the second half of the 14th century. Dating of the different parts of this site was based on historical data, stratigraphy, the study of 7,059 ceramic fragments, and 14C dating.
We collected 10 teeth from three skeletons in Sens, 4 teeth from three skeletons in Dreux, and 5 teeth from two skeletons in Montpellier. The teeth were washed thoroughly with sterile phosphate-buffered saline and fractured longitudinally. Powdery remnants of dental pulp were scraped into sterile tubes for DNA extraction, as previously described (19). Seventeen teeth collected from contemporary dental patients in Marseille without any evidence of plague were used as negative control teeth ("negative teeth").
Amplification of Ancient Y. pestis
Appendix Figure 2
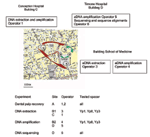
Appendix Figure 2. Detailed experimental process for ancient material to avoid Yersinia pestis contamination.
MST was applied to DNA extracted from 19 teeth from the remains of eight persons in one Justinian era burial and two graves from the time of the second pandemic, as described above. We instituted precautions to avoid bacteriologic and molecular contamination of ancient material (Appendix Figure 2). Two successive experiments were conducted. Dental pulp was recovered in a Y. pestis–free building A by two operators (operators 1 and 2), who had never worked with Y. pestis, its DNA, or amplicons. Teeth were processed individually by cleaning and opening the tooth and recovering the dental pulp into a sterile tube, which was closed securely. Each tooth was processed separately in chronologic order from Justinian to Black Death material, and negative control teeth were processed at the end. New sterile forceps were used for every tooth. Dental pulps were then transported into another laboratory, B1, 200 m away from building A, and to building C, 600 m away from building A for DNA extraction. It was performed by operator 3, who also had never worked with Y. pestis. DNA extraction for each experiment was performed with new reagents, ordered directly from the distributors by operators 1 and 3, who followed the protocol for good practices for ancient DNA manipulation (20). Extracted DNA was submitted to a second laboratory in building B2/D for suicide PCRs (18), which were conducted with one negative control for every three specimens. Finally, amplicons were transported to building D for cloning and sequencing, which was carried out by operators 4 to 6, who had never worked with Y. pestis. PCR products were cloned in PGEM-T Easy Vector (Promega, Charbonnières, France), as described by the manufacturer. Six clones were cultivated in LB medium (USB, Cleveland, OH) overnight, and plasmid purification was performed by using the Promega system. Six clones were sequenced from every amplicon by consensus primers. The sequences were compared with sequences available in GenBank by using the BLAST software version 2.2.8 to ensure accurate species identity. The sequences were further compared by using the BLAST software version 2.2.8 with our local Y. pestis spacer sequence database to ensure proper biovar identity.
Y. pestis Spacer Database
All the isolates were firmly identified as belonging to Y. pestis species on the basis of their phenotypic profile and partial analysis of 16S rRNA gene and rpoB gene sequences. A total of eight intergenic spacers located throughout Y. pestis genome (Table 1) yielded 2–9 alleles per spacer, resulting in a total of 23 molecular events potentially corresponding to >8 x 106 combinations (223 molecular events). The 387-bp spacer YP1 exhibited a 183-bp deletion specific for the Medievalis isolates. For spacer YP3, we observed five alleles: Orientalis isolates 1513 and 695 exhibited a complete, 340-bp sequence, and the other Orientalis isolates exhibited a 16-bp deletion; Medievalis isolates featured a 48-bp deletion and a mutation G → A at position 135 of the spacer; Antiqua isolates exhibited a 32-bp deletion except for isolate 611, which had a 16-bp deletion and mutations C164 → T, C166 → T and A191 → G. Spacer YP3 thus differentiated every biovar. As for spacer YP4, Medievalis isolates were characterized by a 36-bp deletion, whereas Antiqua and Orientalis isolates exhibited a complete spacer. For spacer YP5, Medievalis isolates exhibited a full-length 292-bp spacer, whereas Antiqua and Orientalis isolates exhibited a 32-bp deletion at position 101 of the spacer. For spacer YP7, a total of nine alleles were found; Antiqua isolates were classified within seven alleles, Medievalis isolates within four alleles, and Orientalis isolates within five alleles. Alleles two and three were found only among Orientalis isolates. A full-length sequence of 330 bp was found for strain 549; the other isolates exhibited deletions. At position 126 in the spacer, strains 643 and 695 exhibited a 56-bp deletion; at position 133, strains 304, 1092, 507 and 571 exhibited a 49-bp deletion; at position 140, strains 611, 685, and 537 exhibited a 42-bp deletion; at position 142, strains 616, 548, 565, 518, 520, 560, 561, and 670 exhibited a 35-bp deletion; at position 149, strains 519, 542, 566, 564, and 1513 exhibited a 28-bp deletion; at position 155, strains 552, 613, 772, and 989 exhibited a 21-bp deletion; at position 162, strains 550, 677, and 1594 exhibited a 14-bp deletion; and at position 169 in the spacer, strains 544, 553, and 545 exhibited a 7-bp deletion. As for spacer YP8, Antiqua and Medievalis isolates exhibited a full-length 236-bp spacer, whereas Orientalis isolates had an 18-bp deletion at position 36 of the spacer. As for spacer YP9, Antiqua and Medievalis isolates exhibited a full-length 292-bp sequence; Orientalis isolates had an 18-bp deletion located at position 123 of the spacer. As for YP10 spacer, Y. pestis CO92 strain had a unique sequence of 369-bp; all the other isolates exhibited an 18-bp deletion located at position 177 of the spacer. Sequences herein determined were deposited in GenBank (Appendix 1).
Phylogenetic Analysis
Figure 1
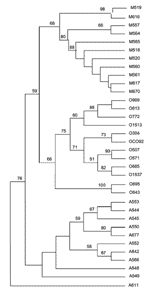
Figure 1. Unrooted tree showing the phylogenetic relationships among the 35 studied Yersinia pestis isolates inferred from sequence analysis of the combination of the eight intergenic spacers using the unweighted pair group method...
Appendix Figure 3

Appendix Figure 3. Unrooted trees showed the phylogenetic relationships among the 35 studied Yersinia pestis isolates inferred from sequence analysis of the combination of the eight intergenic spacers by using the parsimony (A), neighbor-joining...
Among the 35 studied Y. pestis strains, we identified three main phylogenetic clusters, each of which included only strains from a single biovar, i.e., Orientalis, Medievalis, or Antiqua, as observed in the unrooted dendrogram (Figure 1). The same topology was obtained when data were analyzed by parsimony, neighbor-joining, and maximum likelihood analyses (Appendix Figure 3). Within the Medievalis cluster, strains 519 and 616, and strains 557 and 564 were grouped by pairs; no other subgroup was identified. Within the Orientalis cluster, three groups were identified; one group included strains 989, 613, 772, and 1513; a second group was made up of three pairs of strains, i.e., 304 and CO92, 507 and 571, and 685 and 1537; the third group diverged before the differentiation of the other two groups and contained strains 695 and 643. Within the Antiqua cluster, two groups were identified; one group included strains 553, 544, 545, 550, and 677, with the last two clustered; the second group comprised strains 552, 542, and 566; strains 548 and 549 did not group with either of these two groups but diverged before the differentiation of the other Antiqua strains. The Antiqua strain 611 exhibited a unique phylogenetic position. This strain differed from all other studied Y. pestis strains and appeared to have diverged before the separation of the three main clusters.
MST of Ancient Dental Pulp Specimens
Figure 2
![Thumbnail of Molecular detection of Yersinia pestis was achieved in the dental pulp of remains of humans excavated from one Justinian and two Black Death mass graves in France by spacer amplification and sequencing (+, positive polymerase chain reaction [PCR] amplification and sequencing; –, absence of PCR amplification; ND, not done). Sequence analyses showed strains were of Orientalis genotype in all sets of remains; one of them exhibited two mutations numbered according to Y. pestis CO92 stra](/eid/images/03-0933-F2-tn.gif)
Figure 2. Molecular detection of Yersinia pestis was achieved in the dental pulp of remains of humans excavated from one Justinian and two Black Death mass graves in France by spacer amplification and...
In the 46 PCR experiments we performed on ancient tooth samples, we obtained 10 Y. pestis sequences (Figure 2); no sequences were found in the 51 PCR experiments with control teeth (p < 10–4). The teeth from 7 of the 8 persons' remains yielded 10 specific sequences, 3 persons were positive for two molecular targets, but none of the teeth of 17 persons used as negative controls yielded specific sequences (p < 10–4). YP1 PCR yielded an amplicon in one of six tested persons, YP8 PCR yielded an amplicon with identical sequence in six of six tested persons, and YP3 PCR yielded an amplicon in three of seven tested persons. When compared with GenBank database (Appendix 2), theYP1 sequence obtained in skeleton 5 yielded complete similarity with the homologous region in Y. pestis C092 strain over 390 positions and 99% sequence similarity with the homologous region in Y. pestis KIM strain over 174 positions; the YP8 sequence obtained in the six persons yielded 99% sequence similarity with homologous region in Y. pestis C092 strain over 178 positions and complete sequence identity with homologous region in Y. pestis KIM strain over 116 positions; the YP3 sequence obtained in skeletons 4 and 8 yielded complete sequence identity with that of homologous region in Y. pestis strain CO92 over 364 positions and 98% sequence similarity with homologous region in Y. pestis KIM strain over 206 positions; the YP3 sequence obtained in human remain 2 yielded a 98% similarity with homologous region in Y. pestis CO92 strain over 283 positions and a 95% similarity with homologous region in Y. pestis KIM strain over 162 positions. For each one of these 10 amplicons, further matches dropped to <90% sequence similarity over short sequences of 10 nt to 50 nt. When blasted to our local Y. pestis spacer sequence database, the YP1 spacer sequence obtained in human skeleton 5 was identical to that of the Orientalis and Antiqua reference sequences (Appendix 3), whereas smaller BLAST scores were obtained for the Medievalis reference sequences. Regarding the six YP8 spacer sequences obtained in skeletons 1–6, the 12 first best scores were obtained with Orientalis reference sequences. For the YP3 spacer sequence obtained in skeletons 4 and 8, the 10 first best scores were obtained with Orientalis reference sequences. For the YP3 spacer sequence obtained in skeleton 2, the 10 first best scores were obtained with Orientalis reference sequences, and this amplicon exhibited two specific nucleotide substitutions (Figure 2; Appendix 2). These mutations were consistently obtained in six clones.
Our data show that MST differentiates the three biovars of Y. pestis in a collection of 35 isolates representative of the three biovars and originating from various sources and 13 countries. This finding suggests that MST data can be extrapolated to the entire Y. pestis species. Pulsed-field gel electrophoresis (PFGE) has been the only other technique that allows for biovar and strain differentiation, but it requires large amounts of cultured microorganisms, and the stability of PFGE profiles in subculture has been questioned (9). Ribotyping did not classify isolates into their respective biovars (9,21,22). Specific insertion sequences (IS), including IS100 (23), IS285 (24,25), and IS1541 (26,27), were used as markers in restriction fragment length polymorphism (RFLP) analyses (8,28) and in PCR-based technique (29). The last approach produced identical patterns of IS100 distribution in Antiqua and Medievalis isolates (29). A variable-number tandem repeat technique (30,31) had a greater discrimination capacity than did ribotyping, but isolates from different areas were found to harbor identical types. Sequencing of fragments of five housekeeping genes in 36 Y. pestis isolates from various locations and from 12 to 13 isolates from Y. pseudotuberculosis and Y. enterocolitica did not show diversity in any Y. pestis housekeeping gene (8). Indeed, 19 of these 36 Y. pestis isolates were also included in the present work and featured 12 different MST profiles, thus demonstrating the validity of our hypothesis and the usefulness of MST for Y. pestis genotyping. Morever, its format is applicable to microbial analyses of ancient samples since it requires small amounts of DNA and targets small genomic fragments.
Few studies have aimed to disclose intraspecifc phylogenetic relationships of Y. pestis isolates. RFLP, probed with the IS100, indicated that, among 36 Y. pestis isolates, the three biovars formed distinct branches of the phylogenetic tree and that Y. pestis was a clone that evolved from Y. pseudotuberculosis 1,500–20,000 years ago (8). Isolates of biovars Antiqua and Medievalis clustered altogether apart from those belonging to biovar Orientalis. Likewise, a dendrogram constructed by the UPGMA clustering method on a PCR-based IS100 fingerprint database clearly discriminated Y. pestis isolates of the Orientalis biovar that formed a homogeneous group, whereas isolates of the Antiqua and Medievalis biovars mixed together (29). Isolates of biovar Antiqua showed a variety of fingerprinting profiles, whereas Medievalis isolates clustered with the Antiqua isolates originating from Southeast Asia, which suggests their close phylogenetic relationships. In this study, one isolate (strain Nicholisk 51) displayed a genotyping pattern typical of biovar Orientalis isolates, although this isolate was biovar Antiqua and lacked a 93-bp deletion within the glycerol-3-phosphate dehydrogenase glpD gene characteristic for the glycerol-negative Orientalis biovar. Our data support the view that most Y. pestis isolates cluster according to their biovar, but isolates of biovar Antiqua are more distantly related to other isolates than biovars Orientalis and Medievalis are. MST-based phylogenetic reconstructions unexpectedly found that one Y. pestis Antiqua isolate, number 611, formed a fourth branch, which suggests that Y. pestis may comprise four different lineages instead of the three that have been recognized so far. This unique isolate had not been included in Achtman and collaborators' study (8).
MST was applied to ancient human specimens to test the hypothesis that three biovars were responsible for the three historical pandemics. Contamination of ancient samples by modern Y. pestis DNA and cross-contamination were prevented in our experiments. Indeed, we carried out two independent experiments, each with new reagents, in a laboratory where Y. pestis had never been introduced or studied, without positive controls (17).
Appendix Figure 4
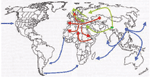
Appendix Figure 4. Geographic origin and routes of spreading of three historical plague pandemics labeled in red (Justinian plague), green (Black Death), and blue (modern), according to historical transcriptions reviewed in Perry (
Both experiments produced consistent results, and negative controls were always negative. That we obtained a unique YP3 sequence further excludes the possibility of contamination, since this sequence differs from all the currently known sequences. This unique sequence was consistently found in six of six clones and thus did not result from the false incorporation of nucleotides by the DNA polymerase. In our previous work on the Black Death, we also reported a unique sequence (17). In the present study, the accurate identification of Y. pestis was confirmed by using two successive sequence analyses. We first blasted the sequences derived from ancient specimens against the GenBank database and observed best matches for Y. pestis homologous sequences, thus ensuring accurate species identification of the amplicons. We then blasted these sequences against our local Y. pestis spacer sequence database and found best matches for homologous sequence in Orientalis reference isolates for the three tested spacers. As our local database has 35 different reference sequences representative of the three Y. pestis biovars, there is no doubt regarding the identification nor the fact that Orientalis-like Y. pestis alone was implicated in the personal remains that we investigated. DNA of Y. pestis was recovered from remains of persons in one mass grave established to be of the Justinian pandemic era on the basis of radiocarbon dating (Appendix Figure 4). We found that the genotype Orientalis, which now occurs worldwide, was involved in all three pandemics. Also, we detected Y. pestis in additonal human remains from Black Death sites, which adds more evidence for its role in the second pandemic in southern France (four sites tested positive) (18,19). Indeed, historical descriptions were suggestive of bubonic plague in medieval southern Europe; in northern Europe, historical data indicated that the Black Death had a different epidemiologic pattern. This finding may indicate that latter outbreaks in the north were not caused by transmission of Y. pestis by blocked rat fleas but rather by mechanical transmission of plague bacteria by another ectoparasite that used humans as their primary hosts. Alternatively, another pathogen may have caused these outbreaks, and a search for Y. pestis in the dental pulps of suspected plague victims in Copenhagen (two persons' remains) and Verdun (five persons' remains) dating from the 18th century failed to show Y. pestis DNA (32). Further studies may elucidate the respective role of Y. pestis and other pathogens that may have contributed to deaths in these times.
Dr. Drancourt is professor of microbiology at Marseille Medical School. His specialties are molecular identification and paleomicrobiology.
References
- Kortepeter M, Parker G. Potential biological weapons threats. Emerg Infect Dis. 1999;5:523–7. DOIPubMedGoogle Scholar
- Perry RD, Fetherston JD. Yersinia pestis–etiologic agent of plague. Clin Microbiol Rev. 1997;10:35–66.PubMedGoogle Scholar
- Wren BW. The yersiniae—a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol. 2003;1:55–64. DOIPubMedGoogle Scholar
- Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, Prentice MB, Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–7. DOIPubMedGoogle Scholar
- Deng W, Burland V, Plunkett G III, Boutin A, Mayhew GF, Liss P, Genome sequence of Yersinia pestis KIM. J Bacteriol. 2002;184:4601–11. DOIPubMedGoogle Scholar
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–40. DOIPubMedGoogle Scholar
- Ogata H, Audic S, Renesto-Audiffren P, Fournier PE, Barbe V, Samson D, Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science. 2001;293:2093–8. DOIPubMedGoogle Scholar
- Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A. 1999;96:14043–8. DOIPubMedGoogle Scholar
- Guiyoule A, Grimont F, Iteman I, Grimont PA, Lefevre M, Carniel E. Plague pandemics investigated by ribotyping of Yersinia pestis strains. J Clin Microbiol. 1994;32:634–41.PubMedGoogle Scholar
- Mollet C, Drancourt M, Raoult D. rpoB sequence analysis as a novel basis for bacterial identification. Mol Microbiol. 1997;26:1005–11. DOIPubMedGoogle Scholar
- Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002;30:42–6. DOIPubMedGoogle Scholar
- Rozen S, Skalestky HJ. Totowa (NJ): Humana Press; 2000. p. 365–86.
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. DOIPubMedGoogle Scholar
- Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA 2: molecular evolutionary genetics analysis software, 2001. Tempe (AZ): Arizona State University; 2001.
- Felsenstein J. PHYLIP–Phylogeny Inference Package (version 3.2). Cladistics. 1989;5:164–6.
- Castex D, Friess M. In: Kemkes-Grottenthaler A, Henke W, editors. Pein und Plagen. Aspekte einer Historischen Epidemiologie. Francfort: Archaea, Epidemiologie; 1998.
- Cabezuelo U, Castex D. Dolmens, sarcophages et pierres tombales. Chartres: Maison de l'Archéologie;1994. p. 68–70.
- Raoult D, Aboudharam G, Crubezy E, Larrouy G, Ludes B, Drancourt M. Molecular indentification by "suicide PCR" of Yersinia pestis as the agent of medieval Black Death. Proc Natl Acad Sci U S A. 2000;97:12800–3. DOIPubMedGoogle Scholar
- Drancourt M, Aboudharam G, Signoli M, Dutour O, Raoult D. Detection of 400-year-old Yersinia pestis DNA in human dental pulp: an approach to the diagnosis of ancient septicemia. Proc Natl Acad Sci U S A. 1998;95:12637–40. DOIPubMedGoogle Scholar
- Golenberg EM. Ancient DNA. New York: Springer-Velag; 1994. p. 237–56.
- Guiyoule A, Rasoamanana B, Buchrieser C, Michel P, Chanteau S, Carniel E. Recent emergence of new variants of Yersinia pestis in Madagascar. J Clin Microbiol. 1997;35:2826–33.PubMedGoogle Scholar
- Lucier TS, Brubaker RR. Determination of genome size, macrorestriction pattern polymorphism, and nonpigmentation-specific deletion in Yersinia pestis by pulsed-field gel electrophoresis. J Bacteriol. 1992;174:2078–86.PubMedGoogle Scholar
- Portnoy DA, Falkow S. Virulence-associated plasmids from Yersinia enterocolitica and Yersinia pestis. J Bacteriol. 1981;148:877–83.PubMedGoogle Scholar
- Protsenko OA, Filippov AA, Kutyrev VV. Integration of the plasmid encoding the synthesis of capsular antigen and murine toxin into Yersinia pestis chromosome. Microb Pathog. 1991;11:123–8. DOIPubMedGoogle Scholar
- Filippov AA, Oleinikov PV, Motin VL, Protsenko OA, Smirnov GB. Sequencing of two Yersinia pestis IS elements, IS285 and IS100. Contrib Microbiol Immunol. 1995;13:306–9.PubMedGoogle Scholar
- Simonet M, Riot B, Fortineau N, Berche P. Invasin production by Yersinia pestis is abolished by insertion of an IS200-like element within the inv gene. Infect Immun. 1996;64:375–9.PubMedGoogle Scholar
- Odaert M, Devalckenaere A, Trieu-Cuot P, Simonet M. Molecular characterization of IS1541 insertions in the genome of Yersinia pestis. J Bacteriol. 1998;180:178–81.PubMedGoogle Scholar
- McDonough KA, Hare JM. Homology with a repeated Yersinia pestis DNA sequence IS100 correlates with pesticin sensitivity in Yersinia pseudotuberculosis. J Bacteriol. 1997;179:2081–5.PubMedGoogle Scholar
- Motin VL, Georgescu AM, Elliott JM, Hu P, Worsham PL, Ott LL, Genetic variability of Yersinia pestis isolates as predicted by PCR-based IS100 genotyping and analysis of structural genes encoding glycerol-3-phosphate dehydrogenase (glpD). J Bacteriol. 2002;184:1019–27. DOIPubMedGoogle Scholar
- Klevytska AM, Price LB, Schupp JM, Worsham PL, Wong J, Keim P. Identification and characterization of variable-number tandem repeats in the Yersinia pestis genome. J Clin Microbiol. 2001;39:3179–85. DOIPubMedGoogle Scholar
- Adair DM, Worsham PL, Hill KK, Klevytska AM, Jackson PJ, Friedlander AM, Diversity in a variable-number tandem repeat from Yersinia pestis. J Clin Microbiol. 2000;38:1516–9.PubMedGoogle Scholar
- Gilbert MT, Cuccui J, White W, Lynnerup N, Titball RW, Cooper A, Absence of Yersinia pestis-specific DNA in human teeth from five European excavations of putative plague victims. Microbiology. 2004;150:341–54. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 10, Number 9—September 2004
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Didier Raoult, Unité des Rickettsies, IFR 48, Faculté de Médecine, Université de la Méditerranée, Marseille – France; fax: 0-33-4-91-38-77-72
Top