Volume 13, Number 7—July 2007
Dispatch
Virus Detection and Monitoring of Viral Load in Crimean-Congo Hemorrhagic Fever Virus Patients
Abstract
We developed a real-time reverse transcription–-PCR that detected 1,164 copies/mL of Crimean-Congo hemorrhagic fever virus per milliliter of serum at 95% probability (probit analysis) and was 100% concordant with nested PCR on 63 samples from 31 patients with confirmed infection. Infected patients who died appeared to have higher viral loads; low viral loads correlated with IgG detection.
Crimean-Congo hemorrhagic fever (CCHF) is a tickborne viral zoonosis that occurs widely in Africa, Asia, and Eastern Europe. It is caused by CCHF virus (CCHFV), a segmented, negative-stranded RNA virus belonging to the family Bunyaviridae, genus Nairovirus. CCHF has a fatality rate of ≈30% and a potential for nosocomial spread (1). Early diagnosis of CCHF is important for case management and protection of medical staff.
Diagnostic assays for CCHF include virus culture, antigen-detection enzyme immunoassay (EIA), antibody-detection EIA, and reverse transcription–PCR (RT-PCR) (2). Virus detection is the main diagnostic method in the acute stage of disease, and RT-PCR is most sensitive method of detection. However, because of the remarkable genetic variability among CCHFV strains, all current RT-PCRs either lack sensitivity or focus on the detection of local CCHFV variants only (3–6).
Figure 1
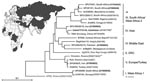
Figure 1. Global distribution and phylogenetic relationships of Crimean-Congo hemorrhagic fever virus (CCHFV) strains selected for design and validation of the assay. All strains except those marked with asterisks were tested. Phylogenetic analysis...
We describe the first real-time RT-PCR that rapidly and reliably detects the global spectrum of clinically relevant virus strains. An extended strategy of probe design was implemented to cover such high variability. Sensitivity was demonstrated by testing virus strain collections from several different Biosafety Level 4 laboratories, essentially covering the full range of global diversity of CCHFV (Figure 1). A comprehensive panel of original clinical samples from persons with confirmed cases of CCHF was used for clinical evaluation; the samples were collected by World Health Organization reference facilities.
Figure 2
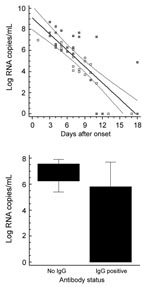
Figure 2. Clinical evaluation. Upper panel: plasma viral load over time in 44 samples from 17 patients. Samples from patients who died are marked with a filled square. Lower panel: plasma viral load...
Primers and probes were selected on the basis of an alignment of S segment sequences of 61 CCHFV isolates from all known CCHF-endemic regions worldwide (7) (representative sequences shown in expanded online version of Figure 2. Oligonucleotide melting points, folding characteristics, and cross-hybridization properties were determined by using the Primer Express software package (Applied Biosystems; Foster City, CA, USA). Primers were selected to amplify a 181-bp region near the 5′-end of the S segment. The capability of these primers to amplify 12 representative CCHFV strains from distinct CCHF-endemic regions was confirmed initially by gel detection RT-PCR (data not shown) (Figure 2, panel B).
For real-time PCR, identifying a simple detection probe compatible with all known CCHFV strains was not possible. Therefore, a broad-range probe was formulated on the basis of the observation that the non–Watson/Crick base pair G:T is almost as thermodynamically stable as regular Watson/Crick base pairs, whereas A:C is very unstable (8). Thus, the probe was placed on the DNA strand that provided more G:T mismatches than complementary A:C mismatches, and the resulting G:T mismatches were not compensated for. As shown in the left panel of Figure 2B this probe, designated SE01, detected all 12 representative strains. Because signal intensity varied according to the strain detected, a strain that provided low signal (BT956, Figure 2B) was chosen for evaluation of sensitivity. Its full S segment RNA was cloned and transcribed in vitro to obtain a quantitative RNA standard (9). Cloning, in vitro transcription, purification, and quantification were performed as previously described (10). End-point dilution showed that single copies of RNA could be detected despite low overall fluorescence (data not shown). Nevertheless, variation in signal intensity between strains was adjusted by the following 2 modifications. First, an additional oligonucleotide (SE03) was introduced at the same binding site as SE01. This probe had 2 effects: first, a pyrimidine base (IUB-code “Y,” 50% C and 50% T) was generated at 2 positions of balanced C/T polymorphisms. Second, a “keto” base (IUB-code “K,” 50% G and 50% T) resulted at 1 position of total variability (A, C, G, T). RNA from the 12 representative strains was tested, and those strains that still provided low signal were realigned separately. On the basis of the second alignment, an additional probe was selected at an alternative binding site to prevent interference with probes at the first binding site. It was placed on the minus strand to obtain more G:T mismatches than complementary A:C mismatches (see above). The improvement obtained by the additional probes on the set of representative strains is shown in Figure 2B, middle panel. The final assay protocol is summarized in the Table.
For precise evaluation of analytical sensitivity, a series of human plasma samples was spiked with the RNA standard from strain BT-958 in concentrations ranging from 100,000 to 10 copies per mL. Testing was done on 5 replicate reactions per concentration, and probit analysis was conducted as shown in the expanded online version of Figure 2, panel B, right graph (11). The calculated limit of detection, defined as the concentration down to which >95% of conducted tests can be expected to be positive, was 13.6 copies per reaction (p = 0.05). This corresponded to 1,164 copies per mL of plasma (95% confidence interval, 780–2990 copies/mL).
Cross-reactivity was excluded by testing DNA or RNA from cultures or high-titered clinical samples containing Dugbe virus, Rift Valley fever virus, Sudan Ebolavirus Gulu, Lassa virus AV, yellow fever virus, dengue virus types 1–4, Japanese encephalitis virus, West Nile virus Uganda, Venezuelan equine encephalitis virus, Sindbis virus, Ross River virus, Epstein-Barr virus, hepatitis C virus, human cytomegalovirus, monkeypox virus, poliomyelitis virus types 1–3, rabies virus RSDD, Bacillus anthracis, Leptospira interrogans, Listeria monocytogenes, Neisseria meningitidis, Coxiella burnetii, Rickettsia prowazekii, R. rickettsii, and Plasmodium falciparum. An additional 128 blood specimens collected during the course of the study from 128 patients with conditions other than CCHF all tested negative for CCHF virus.
The real-time RT-PCR was used to test and quantify 63 serum samples from 31 patients with laboratory-confirmed CCHFV infection; the samples were obtained 1–18 days after symptom onset. All samples had nested RT-PCR results positive for CCHFV (3), and all were also positive by the new real-time RT-PCR. For 21 patients with confirmed CCHF (17 from South Africa, 3 from Iran, and 1 from Pakistan), viral load was quantified and compared with other standard diagnostic methods for CCHFV detection (Appendix Table). Again, sensitivity of the new assay was at least as high as that of nested PCR. As shown in Figure 2, there was a clear correlation between viral load and duration of symptoms in these patients. Clinical outcome could not be correlated clearly with viral load, although patients who died of the disease seemed, in general, to have higher viral loads (Figure 2, filled squares). The appearance of antibodies correlated clearly with lower viral loads (Figure 2).
To our knowledge, this is the first PCR validated with representative CCHFV strains from nearly all regions worldwide where the virus is endemic. High sensitivity enables reliable detection of virus in early stages of the infection, when antibody detection is unreliable or impossible. By eliminating the need for postamplification product processing, real-time RT-PCR enables shortened turnaround times for reporting results, which is critical for deciding on isolation and contact-tracing for suspected case-patients. Quantification of viral load may assist in estimating the patient’s infectivity. It may also assist in predicting the clinical outcome and could be used to monitor viral load in patients receiving ribavirin treatment (12). Our study provides baseline data on CCHF viral load throughout the acute stage of the illness. High viral load tended to indicate fatal outcome, and lower viral load was generally associated with detectable antibodies. Because detectable antibody response correlates with good outcome (13), viral load will probably be a useful predictor of clinical progress. These preliminary data are highly encouraging for further studies on larger patient cohorts.
Dr Wölfel is a medical microbiologist at the Department of Virology and Rickettsiology of the Bundeswehr Institute of Microbiology in Munich, Germany. He is working in the area of medical defense against biological warfare and terrorism, and his interests include viral hemorrhagic fevers and rickettsial diseases.
Acknowledgments
We are grateful to Britta Liedigk, Beate Becker-Ziaja, and John Chamberlain for excellent technical assistance.
This work was supported by the European Commission (contracts SSPE-CT-2003-502567 and SSPE-CT-2005-022639), the Bundeswehr Medical Service (contract E/B41G/1G309/1A403), and the Federal Office of Civil Protection and Disaster Assistance (contract BBK-F-440-00-1).
References
- Hoogstraal H. The epidemiology of tick-borne Crimean-Congo hemorrhagic fever in Asia, Europe, and Africa. J Med Entomol. 1979;15:307–417.PubMedGoogle Scholar
- Burt FJ, Leman PA, Smith JF, Swanepoel R. The use of a reverse transcription–polymerase chain reaction for the detection of viral nucleic acid in the diagnosis of Crimean-Congo haemorrhagic fever. J Virol Methods. 1998;70:129–37. DOIPubMedGoogle Scholar
- Duh D, Saksida A, Petrovec M, Dedushaj I. vsic-Zupanc T. Novel one-step real-time RT-PCR assay for rapid and specific diagnosis of Crimean-Congo hemorrhagic fever encountered in the Balkans. J Virol Methods. 2006;133:175–9. DOIPubMedGoogle Scholar
- Schwarz TF, Nsanze H, Longson M, Nitschko H, Gilch S, Shurie H, Polymerase chain reaction for diagnosis and identification of distinct variants of Crimean-Congo hemorrhagic fever virus in the United Arab Emirates. Am J Trop Med Hyg. 1996;55:190–6.PubMedGoogle Scholar
- Yapar M, Aydogan H, Pahsa A, Besirbellioglu BA, Bodur H, Basustaoglu AC, Rapid and quantitative detection of Crimean-Congo hemorrhagic fever virus by one-step real-time reverse transcriptase-–PCR. Jpn J Infect Dis. 2005;58:358–62.PubMedGoogle Scholar
- Deyde VM, Khristova ML, Rollin PE, Ksiazek TG, Nichol ST. Crimean-Congo hemorrhagic fever virus genomics and global diversity. J Virol. 2006;80:8834–42. DOIPubMedGoogle Scholar
- Peyret N, Seneviratne PA, Allawi HT, SantaLucia J Jr. Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A.A, C.C, G.G, and T.T mismatches. Biochemistry. 1999;38:3468–77. DOIPubMedGoogle Scholar
- Hewson R, Chamberlain J, Mioulet V, Lloyd G, Jamil B, Hasan R, Crimean-Congo haemorrhagic fever virus: sequence analysis of the small RNA segments from a collection of viruses world wide. Virus Res. 2004;102:185–9. DOIPubMedGoogle Scholar
- Drosten C, Gottig S, Schilling S, Asper M, Panning M, Schmitz H, Rapid detection and quantification of RNA of Ebola and Marburg viruses, Lassa virus, Crimean-Congo hemorrhagic fever virus, Rift Valley fever virus, dengue virus, and yellow fever virus by real-time reverse transcription-PCR. J Clin Microbiol. 2002;40:2323–30. DOIPubMedGoogle Scholar
- Fink H, Hund G. Probit analysis with programmed computers [in German]. Arzneimittelforschung. 1965;15:624–30.PubMedGoogle Scholar
- Ergonul O, Celikbas A, Dokuzoguz B, Eren S, Baykam N, Esener H. Characteristics of patients with Crimean-Congo hemorrhagic fever in a recent outbreak in Turkey and impact of oral ribavirin therapy. Clin Infect Dis. 2004;39:284–7. DOIPubMedGoogle Scholar
- Ergonul O, Celikbas A, Baykam N, Eren S, Dokuzoguz B. Analysis of risk-factors among patients with Crimean-Congo haemorrhagic fever virus infection: severity criteria revisited. Clin Microbiol Infect. 2006;12:551–4. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 13, Number 7—July 2007
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Christian Drosten, Bernhard Nocht Institute for Tropical Medicine, Bernhard Nocht Str. 74, 20359 Hamburg, Germany;
Top