Volume 15, Number 9—September 2009
Research
Predicting Phenotype and Emerging Strains among Chlamydia trachomatis Infections
Cite This Article
Citation for Media
Abstract
Chlamydia trachomatis is a global cause of blinding trachoma and sexually transmitted infections (STIs). We used comparative genomics of the family Chlamydiaceae to select conserved housekeeping genes for C. trachomatis multilocus sequencing, characterizing 19 reference and 68 clinical isolates from 6 continental/subcontinental regions. There were 44 sequence types (ST). Identical STs for STI isolates were recovered from different regions, whereas STs for trachoma isolates were restricted by continent. Twenty-nine of 52 alleles had nonuniform distributions of frequencies across regions (p<0.001). Phylogenetic analysis showed 3 disease clusters: invasive lymphogranuloma venereum strains, globally prevalent noninvasive STI strains (ompA genotypes D/Da, E, and F), and nonprevalent STI strains with a trachoma subcluster. Recombinant strains were observed among STI clusters. Single nucleotide polymorphisms (SNPs) were predictive of disease specificity. Multilocus and SNP typing can now be used to detect diverse and emerging C. trachomatis strains for epidemiologic and evolutionary studies of trachoma and STI populations worldwide.
Chlamydia trachomatis is spread by close social contact or sexual activity. Worldwide, C. trachomatis is the leading preventable cause of blindness and bacterial sexually transmitted infections (STIs). Various typing techniques have been developed to better understand the epidemiology and pathogenesis of chlamydial diseases. Early typing schemes used monoclonal and polyclonal antibodies directed against the major outer membrane (MOMP) (1), and differentiated the organism into serovars and seroclasses: B class (comprising serovars B, Ba, D, Da, E, L2, L2a), C class (A, C, H, I, Ia, J, Ja, K, L1, L3), and intermediate class (F, G). Sequencing of ompA, which encodes MOMP, has refined typing, detecting numerous trachoma (2,3) and sexually transmitted (4,5) serovar subtypes.
Seroclasses, however, do not correlate with disease phenotypes. For example, A, B, Ba, and C are responsible for trachoma, whereas lymphogranuloma venereum (LGV) strains, L1-3, are associated with invasive diseases, such as suppurative lymphadenitis and hemorrhagic proctitis (6). Other typing techniques, such as restriction fragment length polymorphism ( (7), random amplification of polymorphic DNA, or pulsed-field gel electrophoresis (PFGE) (8), and amplified fragment length polymorphism (AFLP) (9) also correlate poorly with disease phenotype, and none have been standardized across laboratories.
Multilocus sequence typing (MLST) has been used to characterize strains and lineages of numerous pathogens associated with human diseases that cause serious illness and death, including Neisseria meningitidis, Staphylococcus aureus, Vibrio cholerae, and Haemophilus influenzae. MLST uses 500–700 bp sequences of internal regions of 6–8 housekeeping genes, excluding genes suspected to be under immune selection (where there is positive selection for sequence diversity) and ribosomal RNA genes (which are multicopy and too conserved) (10). Advantages of MLST include its precision, allowing simple interlaboratory comparisons, good discrimination between strains, and buffering against the distorting effect of recombination on genetic relatedness. MLST data are also amenable to various population genetic analyses (11,12). Databases for >30 species are curated at www.mlst.net and pubmlst.org. In parallel with our study, 2 multilocus schemes have recently been developed for C. trachomatis. The first violated the above premise by including ompA, which is under immune selection (13). The second included only laboratory-adapted and 5 clinical E strains from the Netherlands (14).
In this work, unlike the other C. trachomatis MLST schemes, we used complete genomic comparisons of 7 strains from 4 species within the family Chlamydiaceae to identify conserved candidate housekeeping genes across the genomes. This approach ensures that the chosen loci are stable over the course of evolution, and allows for future application of a unified MLST scheme for other Chlamydiaceae spp. We typed a diverse worldwide collection of reference and clinical isolates from trachoma and STI populations, correlating genetic variation with geography and disease phenotype. We found disease-specific single nucleotide polymorphisms (SNPs) and a diversity of new strains including recombinant strains that occurred for ompA relative to housekeeping loci, following up on our recent discovery of this phenomenon at multiple loci in Chlamydiaceae genomes (15–17).
Reference and Clinical Samples
Nineteen C. trachomatis reference strains (A/SA-1, B/TW-5, Ba/Apache-2, C/TW-3, D/UW-3, Da/TW-448, E/Bour, F/IC-Cal3, G/UW-57, H/UW-4, I/UW-12, Ia/IU-4168, J/UW-36, Ja/UW-92, K/UW-31, L1/440, L2/434, L2a/TW-396, and L3/404) and 68 clinical isolates from 6 geographic locations worldwide (obtained from patients with trachoma and STIs including proctitis) were analyzed. Because de-identified clinical data and samples were used, the research was considered institutional review board exempt by Children’s Hospital Oakland Research Institute.
Selection of Housekeeping Genes
Figure 1

Figure 1. Comparison of 14 housekeeping genes among genome sequences of 4 Chlamydiaceae species and 7 strains. Circle 1, genes on forward Chlamydia trachomatis strand, color coded by role category; Circle 2, genes on...
We genome-sequenced 7 strains from 4 species of the 2 genera of Chlamydiaceae: C. trachomatis (strains D/UW-3/CX [18] and A/Har-13 [19]), Chlamydia muridarum (rodent strain MoPn [20]), Chlamydophila pneumoniae (human strains AR39 [20]; CWL029 [21], and J138 [21]), and Chlamydophila caviae (guinea pig inclusion conjunctivitis strain [22]), the most distantly related species of Chlamydiaceae. On the basis of comparative genomics (20) and comparisons generated by CGView (23), we identified an initial candidate pool of 14 housekeeping genes (Figure 1) present in all 7 genomes with an average BLAST score ratio (BSR) (24) >0.5 for orthologs queried against C. caviae relative to the BLAST score of each sequence against itself. The BSR of >0.5 provides a cutoff to select genes that have lower levels of nucleotide sequence divergence in the genome (i.e., putative housekeeping genes). We then selected 7 genes (Figure 1) on the basis of i) diverse chromosomal regions where a single recombinational exchange would be unlikely to co-introduce >1 selected gene; ii) regions where several contiguous genes were involved in metabolic or key functions; iii) essential metabolic enzymes (e.g., tRNA synthases); iv) genes without similarity to human genes; and v) no genes under diversifying selection.
ompA and MLST Analyses
DNA was extracted from isolates using Roche High Pure Kits (Roche Diagnostics, Pleasanton, CA, USA) and ompA genotyped as described (15,16,25). DNASTAR (Madison, WI, USA) was used to design primers to amplify ≈600–700 bp for each gene (Table 1); BLAST (NCBI; http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to ensure primer specificity for C. trachomatis genes. MLST PCR was carried out in 96-well plates as described (26). Sequenced DNA (GenBank accession nos. FJ45414–FJ746022) using ABI3700 instruments were aligned by using MegAlign (DNASTAR). Each unique sequence for a locus was designated as a unique allele using Sequence Output (www.mlst.net). Each allelic profile (made up of the string of integers corresponding to allele numbers at the 7 loci) was assigned as a different strain or clone and given an ST as a clone descriptor. All STs have been deposited in the C. trachomatis site at www.mlst.net.
Allelic profiles and concatenated sequences were used to determine the relatedness of isolates. Average pairwise diversity between isolates was calculated from the 3,714-bp concatenated sequence of the 7 loci for each isolate joined in-frame using MEGA4 (27). Synonymous (dS) and nonsynonymous (dN) substitutions were determined using MEGA4 for each locus. Allele frequencies per locus and geographic region were calculated using SAS software 9.2 (SAS Institute, Inc., Cary, NC, USA) with the PROC FREQ tool supplying the frequency count. We calculated a classification index (11) on the basis of allele and ST frequency between populations of different geographic regions to determine the probability of association of an allele with a particular continental/subcontinent region. Statistical significance was determined by 10,000 resamplings of allele and ST frequency per region.
Strain Clustering and Single Nucleotide Polymorphism Analyses
eBURST (www.mlst.net) was used to identify clusters of related and singleton STs that were not closely related to any other ST (12) and to predict patterns of evolutionary descent. MEGA4 (27 )was used to construct a tree from concatenated sequences by using minimum evolution, neighbor joining, or unweighted pair group method with arithmetic mean, with various substitution models including Kimura 2-parameter, Jukes Cantor, and p-distance; 1,000 bootstrap replicates were used to test support for each node in the tree. The short evolutionary distances (<≈0.01) imply that back-substitutions were rare, and as expected, all methods gave similar results (data not shown). SplitsTree (www.splitstree.org) was used for evolutionary tree construction by decomposition analyses using the distance matrix produced from pairwise comparisons of concatenated sequences to determine interconnected networks (28).
A matrix of all SNPs by ST was produced in Excel. SAS was used to identify which SNPs were associated with an ST using PROC FREQ. Statistical significance was determined by using a classification index as above for the probability of association of a SNP with a particular ST. Levene’s test (29) was used to determine whether there was equal variance across the 87 isolates. A p value of <0.05 was considered significant.
Discrimination of C. trachomatis by MLST
Figure 1 shows genomic alignments for C. trachomatis (D/UW-3/CX), C. muridarum, C. pneumoniae (AR39), and C. caviae. The C. trachomatis (A/Har-13 and D/UW-3/CX) and C. pneumoniae (AR39, CWL029, and J138) genome sequences were almost identical within species for gene content and could be represented by D/UW-3/CX and AR39, respectively.
ompA genotypes were compared with STs resolved by MLST. The Technical Appendix shows ompA genotype (first letter of strain ID), ST, assigned alleles, and clinical characteristics for each isolate. There were 44 STs (0.51 ST/isolate) for the 87 isolates. Thirty STs were represented by a single isolate. In some cases, STI isolates from diverse geographic regions shared the same ST. For example, ompA genotype E STI isolates (ST39) were found in California, USA; Amsterdam, the Netherlands; Ecuador; Lisbon, Portugal; and Tanzania. Similarly, LGV genotypes (ST1) were identified in San Francisco, California, USA; Seattle, Washington, USA; and Amsterdam; 2 clinical L2b genotypes (ST33) were restricted to Amsterdam. In contrast, no trachoma isolates from different continents shared the same ST.
ompA genotypes correlated poorly with relatedness between strains by MLST data (Technical Appendix). Isolates of the same ST had up to 4 different ompA genotypes. For example, ST19 included ompA genotypes D, H, I, and J. For each ompA genotype, 38%–100% belonged to different STs. Different STs with the same ompA genotype were closely related by MLST (e.g., isolates with C and F ompA genotypes); others were not. Isolates of D, E, and Ja ompA genotypes differed at as many as 5 MLST loci.
Allele Characteristics and Localization by Geography
Allele characteristics are shown in Table 2. The number of alleles at each locus varied from 4 to 11. The average pairwise distance and dS and dN are provided. We determined allele frequencies on the basis of continental/subcontinental regions (Table 3). The majority of alleles were observed multiple times. Seventeen were found only once, and 28 were unique to a specific region (Table 3). The range was from 1 allele at the lysS locus for South America to 9 in North America. The highest frequency of a unique allele was 84.6% (leuS allele 7) for Asia, which also had the highest proportion of unique alleles, 6/17 (35.29%). There was a significant nonuniform distribution of alleles at each locus by classification index.
Phylogenetic Grouping of STs by Disease Phenotypes and Evidence for Recombination
Figure 2

Figure 2. eBURST population snapshot for Chlamydia trachomatis isolates. Sequence types (STs) that are linked differ at single multilocus sequence typing locus and represent clonal complexes. Strains in the same clonal complexes are...
eBURST (11,12) generated 3 clonal complexes (CC) (Figure 2): trachoma strains, A, B, Ba, and C (CC-A); noninvasive STIs with low population prevalence (CC-B); and noninvasive, globally prevalent D/Da, E, and F STIs (CC-C). The Technical Appendix shows eBURST data, including single, double, and triple locus variants (S/D/TLVs).
Figure 3
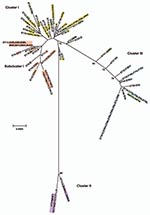
Figure 3. Minimum evolution tree. The tree was constructed using the matrix of pairwise differences between the 87 concatenated sequences for the 7 loci using maximum composite likelihood method for estimating genetic distances....
Relationships between the isolates was further explored by constructing a minimum-evolution tree using MEGA4 (27). These data showed 3 disease clusters (Figure 3). Cluster I comprised noninvasive STIs (eBURST CC-B) and a trachoma subcluster (eBURST CC-A). Cluster II comprised only invasive LGV strains. Cluster III included noninvasive prevalent D/Da, E and F STIs (eBURST CC-C). E58t strain (ST39; cluster III) was isolated from the conjunctiva of a trachoma patient, most likely representing autoinoculation from the urogenital tract, because all other isolates of this ST were from STIs.
Figure 4
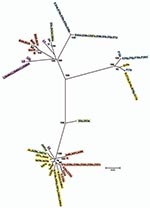
Figure 4. Minimum evolution tree for ompA. The tree was constructed using the matrix of pairwise differences between the 87 sequences by using the maximum composite likelihood method for estimating genetic distances. Numbers...
Nine isolates did not localize on the MLST tree with strains of the same ompA genotype (Figure 3). Ja41nl and Ja47nl, which were expected to cluster with other J and Ja isolates in cluster I if the genome sequences were similar, were identical by MLST to reference strain F and clinical isolates F8p, F9p, E19e, and E5s in cluster III. Similarly, D83s, which were expected to cluster with other D and Da isolates in cluster III, had the same ST as H40nl, H18s, I22p, and J44nl in cluster I; D2s were identical to Ia and Ia57e in cluster I. Additionally, G16p did not cluster with the other G isolates in cluster I. In analyzing locations of incongruence between clinical D and E isolates in cluster I, compared with those in cluster III, the loci that differed were glyA, yhbG, and pykF in which allele assignments were identical, in general, to G, H, I, Ia, J, Ja, and K strains (Technical Appendix) in cluster I. These were the exact same loci that differed for Ja41nl and Ja47nl in cluster III, compared with other J/Ja isolates in cluster I. Ja26s differed at glyA, mdhC, and yhbG, whereas G16p differed at yhbG, lysS, and leuS. Furthermore, the ompA tree (Figure 4) was incongruent with the MLST tree. We interpret all 9 isolates to be recombinants.
Figure 5
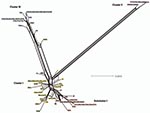
Figure 5. SplitsTree obtained by using concatenated sequences of the 7 loci for the 87 isolates. Cluster I, noninvasive, nonprevalent Chlamydia trachomatis strains (gold) with trachoma Subcluster I (red); cluster II, invasive lymphogranuloma...
SplitsTree decomposition evaluated alternative evolutionary pathways that might indicate recombination between MLST loci (Figure 5). There was considerable network structure, providing evidence of alternative pathways between strains, which may indicate that recombination has influenced the evolution of housekeeping genes for the C. trachomatis strains.
SNPs Associated with Disease Phenotypes
We identified 61 polymorphic sites among the 7 loci. Multiple SNPs were significantly associated with each of the 3 clusters and disease groups (Table 4). For example, 15 SNPs in yhbG and leuS were 100% specific for all LGV strains in cluster III. Any 1 of these SNPs could be used to identify these strains. SNPs 4, 29, 31, 33, and 34 (together or any 1 alone) were specific for the cluster II STIs. For the trachoma Subcluster I, unlike for other clusters, only SNP 38 was associated with reference strains B and C and all clinical trachoma strains; SNPs 54, 55 and 57 together (but not any alone) represented all trachoma strains except reference strain A. Based on classification indices and Levene’s test, the null hypothesis of a uniform distribution of SNPs was rejected at the respective locus.
Accumulating evidence for recombination among Chlamydiaceae in general, and C. trachomatis in particular, has motivated a typing system that provides buffering from the distorting effects of genetic reshuffling that plague systems based on a single locus. We therefore developed an MLST scheme derived from comparative genomics of species within the family Chlamydiaceae to select conserved chromosomally dispersed housekeeping genes. Our scheme showed considerable variability in allelic profiles associated with geographic regions, as well as diverse and recombinant strains. We also identified SNPs that correlated with the 3 C. trachomatis disease groups: invasive LGV diseases, noninvasive urogenital diseases, and trachoma.
Comparative genomics of Chlamydia and Chlamydophila spp. identified 14 conserved housekeeping genes that could be used to extend MLST schemes for these and potentially other Chlamydiaceae spp. Surprisingly, each gene was located in a different position within the respective genome, indicating a lack of synteny among chromosomes (20) (Figure 1), except for the 2 C. trachomatis and 3 C. pneumoniae strains, which share within species >99% nucleotide sequence identity. This finding suggests that future schemes should select loci to ensure reasonable coverage of the chromosome.
Although there was relatively little sequence diversity in the housekeeping genes, the number of STs (0.51 ST/isolate) was similar to that of other bacterial pathogens. The previous C. trachomatis MLST scheme had 0.60 ST/isolate (14). None of the loci were identical to ours. In a recent study of the bacterium Burkholderia pseudomallei in Australia, there were 0.65 ST/isolate (11,12) with relatively little diversity and few alleles per locus. However, high levels of recombination are believed to shuffle alleles to generate different large numbers of allelic profiles (STs). The extent to which recombination among alleles generates novel STs in C. trachomatis is unclear. Although the number of STs per isolate varies, the majority of MLST schemes have been successful for strain discrimination, epidemiologic studies, and evaluation of organism evolution (10). MLST, however, may not be sufficiently discriminatory for some epidemiologic investigations, even with increased loci numbers. This may be the case for LGV strains, although our scheme resolved 2 L2b strains from all other LGV strains.
We found that a number of STs for STI isolates were shared across continents. This finding was particularly evident for those from Amsterdam, Ecuador, Lisbon, and San Francisco, which would be expected given increasing opportunities for global travel and international sexual encounters. Notably, L2b isolates (ST33) from proctitis cases differed at 2 loci from other LGV isolates (ST1) and were restricted to Amsterdam. Although some L2b strains from Amsterdam and San Francisco have historically been similar (30), ST differentiation most likely reflects the emergence of these strains among men who have sex with men. Not surprisingly, STs for trachoma isolates were restricted to the geographic region of origin where populations travel only locally.
Allele frequencies were assigned on the basis of continental/subcontinental regions (Table 3). Most alleles were observed multiple times, and more than half were region specific. Despite the opportunity for worldwide spread, some strains may be stable within the respective geographic populations. This stability was particularly evident in Africa and Asia, where the frequency of unique alleles was the highest, although this finding also reflects the fact that most isolates were from trachoma populations. As expected, we found, in general, a statistically significant nonuniform distribution of alleles.
Analyses using eBURST and trees constructed in MEGA4 resolved isolates into clonal complexes or clusters. Both methods identified distinctive groupings of strains by disease phenotypes. STIs caused by less common strains formed an eBURST group (CC-B) but were within cluster I on the tree together with the trachoma Subcluster I, which was a separate eBURST group (CC-A). A similar clustering pattern to our tree was found by Pannyhoek et al. (14) by using 16 reference and 5 clinical E strains, but they did not distinguish trachoma reference strain B/TW-5 from the LGV group. Our cluster II included only LGV strains. Cluster III contained the noninvasive globally prevalent D/Da, E, and F strains (eBURST CC-C). This cluster represents efficiently transmitted strains with adaptive fitness in the genital tract.
A number of isolates representing different ompA genotypes shared the same ST, whereas many isolates of the same ompA genotype had different STs (Technical Appendix). Furthermore, 9 isolates were found outside the expected cluster, suggesting that recombinational replacement at the ompA locus occurs relatively frequently. Accumulating evidence supports frequent recombination among Chlamydiaceae. Initial evidence came from observations of recombination within ompA (4,31) followed by phylogenetic analyses (32), and bioinformatic and statistical analyses for multiple species of the family Chlamydiaceae and C. trachomatis strains (15). Recently, we showed intergenic recombination involving ompA and pmpC, pmpE-I, and frequent recombination throughout the genome with significant hotspots for recombination for recent clinical isolates (16,17). Pannekoek et al. noted incongruence between ompA and fumC sequences (14). Most recombination in our study involved yhbG, glyA, and pykF (Technical Appendix) with incongruence, compared with ompA. Based on C. trachomatis recombinants that have been created in vitro, the estimated size of transferred DNA ranged from 123 kb to 790 kb (33). Although additional recombination sites may exist in regions that were not sequenced, any gene in our study could be involved in lateral gene exchange with a range of 1,191 bp for a single gene (e.g., ompA), 27 kb (yhbG to ompA) to at least 248 kb (glyA to yhbG), which is consistent with DeMars and Weinfurter (33) and our previous findings (16,17).
Analysis of the 61 SNPs among the 7 loci showed a statistically significant association of specific polymorphisms with each disease cluster (Table 4). A total of 15 SNPs singly or together identified the LGV cluster. Similarly, 5 SNPs identified the prevalent cluster II D/Da, E, and F strains. Three clinical D and E strains did not contain these SNPs and each appeared to be a recombinant with other STI strains. Only 1 SNP (in pykF) identified all trachoma strains in Subcluster I. Reference trachoma strains A and Ba did not contain this SNP, suggesting that they may not represent circulating strains among present-day populations.
Other studies have associated SNPs or indels in pmp and porB genes with specific disease causing C. trachomatis clades (16,34,35). However, SNPs were not individually analyzed for specific disease associations and the target genes encode surface exposed proteins likely to be under selection for epitope variation to avoid immune system surveillance. A frame-shift mutation in 1 of the tryptophan synthase genes, trpA, was associated with trachoma strains when compared with all others, although some B and C strains lack the entire gene (35). Large deletions in the cytotoxin loci have also been identified that differentiate the 3 disease groups, yet strain B is missing these loci (36). The latter study relied on reference strains, which may limit the use of these deletions for identifying disease-specific groups because clinical isolates may vary in deletion size or location. Additionally, tryptophan synthase genes and cytotoxin loci are located within the 50-kb plasticity zone of the chromosome, a region known for genetic reshuffling (20). The current study differs from those previously mentioned in that it used housekeeping genes that are not under immune selection or in the plasticity zone. Therefore, the SNPs we identified are probably neutral and can be used as reliable markers for disease association. Furthermore, SNPs were based on reference and clinical isolates of multiples of the same strains from 6 geographic regions, representing a broad diversity of this species.
Given the high rates of infection among STI (37) and trachoma populations (38,39), the ability to distinguish LGV and noninvasive urogenital and trachoma strains, including mixed infections, would aid epidemiologists, clinicians, and public healthcare workers worldwide in determining appropriate therapeutic or intervention strategies (40). Our multilocus and SNP typing can now be used to standardize the way an organism is typed; isolates from diverse geographic regions worldwide can be identified and compared; and diverse and emerging C. trachomatis strains can be detected for epidemiologic and evolutionary studies among trachoma and STI populations worldwide.
Dr Dean is director of Children’s Global Health Initiative, and a faculty member at University of California at San Francisco and the University of California at Berkeley Joint Graduate Group in Bioengineering. She is also a senior scientist in the Center for Immunobiology and Vaccine Development at Children’s Hospital Oakland Research Institute. Her research interests focus on chlamydial pathogenesis and comparative genomics and molecular epidemiology of chlamydial ocular and sexually transmitted diseases.
Acknowledgments
We thank Hugh Taylor for providing trachoma samples from Tanzania.
This work was supported by NIH grants R01 AI059647, R01 AI039499 and R01 EY/AI012219 (D.D.), a European Union grant EU-FP6-LSHG-CT-2007-037637 (S.A.M.), and a Wellcome Trust Grant 030662 (B.G.S.).
References
- Wang SP, Grayston JT. Three new serovars of Chlamydia trachomatis: Da, Ia, and L2a. J Infect Dis. 1991;163:403–5.PubMedGoogle Scholar
- Dean D, Schachter J, Dawson CR, Stephens RS. Comparison of the major outer membrane protein variant sequence regions of B/Ba isolates: a molecular epidemiologic approach to Chlamydia trachomatis infections. J Infect Dis. 1992;166:383–92.PubMedGoogle Scholar
- Hayes LJ, Bailey RL, Mabey DC, Clarke IN, Pickett MA, Watt PJ, Genotyping of Chlamydia trachomatis from a trachoma-endemic village in the Gambia by a nested polymerase chain reaction: identification of strain variants. J Infect Dis. 1992;166:1173–7.PubMedGoogle Scholar
- Brunham R, Yang C, Maclean I, Kimani J, Maitha G, Plummer F. Chlamydia trachomatis from individuals in a sexually transmitted diseases core group exhibit frequent sequence variation in the major outer membrane protein (omp1) gene. J Clin Invest. 1994;94:458–63. DOIPubMedGoogle Scholar
- Dean D, Oudens E, Bolan G, Padian N, Schachter J. Major outer membrane protein variants of Chlamydia trachomatis are associated with severe upper genital tract infections and histopathology in San Francisco. J Infect Dis. 1995;172:1013–22.PubMedGoogle Scholar
- Hamill M, Benn P, Carder C, Copas A, Ward H, Ison C, The clinical manifestations of anorectal infection with lymphogranuloma venereum (LGV) versus non-LGV strains of Chlamydia trachomatis: a case-control study in homosexual men. Int J STD AIDS. 2007;18:472–5. DOIPubMedGoogle Scholar
- Frost EH, Deslandes S, Veilleux S, Bourgaux-Ramoisy D. Typing Chlamydia trachomatis by detection of restriction fragment length polymorphism in the gene encoding the major outer membrane protein. J Infect Dis. 1991;163:1103–7.PubMedGoogle Scholar
- Rodriguez P, Allardet-Servent A, de Barbeyrac B, Ramuz M, Bebear C. Genetic variability among Chlamydia trachomatis reference and clinical strains analyzed by pulsed-field gel electrophoresis. J Clin Microbiol. 1994;32:2921–8.PubMedGoogle Scholar
- Morré SA, Ossewaarde JM, Savelkoul PH, Stoof J, Meijer CJ, van den Brule AJ. Analysis of genetic heterogeneity in Chlamydia trachomatis clinical isolates of serovars D, E, and F by amplified fragment length polymorphism. J Clin Microbiol. 2000;38:3463–6.PubMedGoogle Scholar
- Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. 2006;60:561–88. DOIPubMedGoogle Scholar
- Jolley KA, Wilson DJ, Kriz P, McVean G, Maiden MC. The influence of mutation, recombination, population history, and selection on patterns of genetic diversity in Neisseria meningitidis. Mol Biol Evol. 2005;22:562–9. DOIPubMedGoogle Scholar
- Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–30. DOIPubMedGoogle Scholar
- Klint M, Fuxelius HH, Goldkuhl RR, Skarin H, Rutemark C, Andersson SG, High-resolution genotyping of Chlamydia trachomatis strains by multilocus sequence analysis. J Clin Microbiol. 2007;45:1410–4. DOIPubMedGoogle Scholar
- Pannekoek Y, Morelli G, Kusecek B, Morre SA, Ossewaarde JM, Langerak AA, Multi locus sequence typing of Chlamydiales: clonal groupings within the obligate intracellular bacteria Chlamydia trachomatis. BMC Microbiol. 2008;8:42. DOIPubMedGoogle Scholar
- Millman KL, Tavare S, Dean D. Recombination in the ompA gene but not the omcB gene of Chlamydia contributes to serovar-specific differences in tissue tropism, immune surveillance, and persistence of the organism. J Bacteriol. 2001;183:5997–6008. DOIPubMedGoogle Scholar
- Gomes JP, Nunes A, Bruno WJ, Borrego MJ, Florindo C, Dean D. Polymorphisms in the nine polymorphic membrane proteins of Chlamydia trachomatis across all serovars: evidence for serovar Da recombination and correlation with tissue tropism. J Bacteriol. 2006;188:275–86. DOIPubMedGoogle Scholar
- Gomes JP, Bruno WJ, Nunes A, Santos N, Florindo C, Borrego MJ, Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 2007;17:50–60. DOIPubMedGoogle Scholar
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–9. DOIPubMedGoogle Scholar
- Carlson JH, Porcella SF, McClarty G, Caldwell HD. Comparative genomic analysis of Chlamydia trachomatis oculotropic and genitotropic strains. Infect Immun. 2005;73:6407–18. DOIPubMedGoogle Scholar
- Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28:1397–406. DOIPubMedGoogle Scholar
- Shirai M, Hirakawa H, Kimoto M, Tabuchi M, Kishi F, Ouchi K, Comparison of whole genome sequences of Chlamydia pneumoniae J138 from Japan and CWL029 from USA. Nucleic Acids Res. 2000;28:2311–4. DOIPubMedGoogle Scholar
- Read TD, Myers GS, Brunham RC, Nelson WC, Paulsen IT, Heidelberg J, Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 2003;31:2134–47. DOIPubMedGoogle Scholar
- Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36:W181-4. DOIPubMedGoogle Scholar
- Rasko DA, Myers GS, Ravel J. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics. 2005;6:2. DOIPubMedGoogle Scholar
- Millman K, Black CM, Johnson RE, Stamm WE, Jones RB, Hook EW, Population-based genetic and evolutionary analysis of Chlamydia trachomatis urogenital strain variation in the United States. J Bacteriol. 2004;186:2457–65. DOIPubMedGoogle Scholar
- Meats E, Feil EJ, Stringer S, Cody AJ, Goldstein R, Kroll JS, Characterization of encapsulated and noncapsulated Haemophilus influenzae and determination of phylogenetic relationships by multilocus sequence typing. J Clin Microbiol. 2003;41:1623–36. DOIPubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–67. DOIPubMedGoogle Scholar
- Levene H. Robust tests for the equality of variances. In: Olkin I, editor. Contributions to probability and statistics: essays in honor of Harold Hotelling. Stanford (CA): Stanford University Press; 1960. p. 278–92.
- Spaargaren J, Schachter J, Moncada J, de Vries HJ, Fennema HS, Pena AS, Slow epidemic of lymphogranuloma venereum L2b strain. Emerg Infect Dis. 2005;11:1787–8.PubMedGoogle Scholar
- Hayes LJ, Yearsley P, Treharne JD, Ballard RA, Fehler GH, Ward ME. Evidence for naturally occurring recombination in the gene encoding the major outer membrane protein of lymphogranuloma venereum isolates of Chlamydia trachomatis. Infect Immun. 1994;62:5659–63.PubMedGoogle Scholar
- Fitch WM, Peterson EM, de la Maza LM. Phylogenetic analysis of the outer-membrane-protein genes of chlamydiae, and its implication for vaccine development. Mol Biol Evol. 1993;10:892–913.PubMedGoogle Scholar
- DeMars R, Weinfurter J. Interstrain gene transfer in Chlamydia trachomatis in vitro: mechanism and significance. J Bacteriol. 2008;190:1605–14. DOIPubMedGoogle Scholar
- Brunelle BW, Sensabaugh GF. The ompA gene in Chlamydia trachomatis differs in phylogeny and rate of evolution from other regions of the genome. Infect Immun. 2006;74:578–85. DOIPubMedGoogle Scholar
- Caldwell HD, Wood H, Crane D, Bailey R, Jones RB, Mabey D, Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J Clin Invest. 2003;111:1757–69.PubMedGoogle Scholar
- Carlson JH, Hughes S, Hogan D, Cieplak G, Sturdevant DE, McClarty G, Polymorphisms in the Chlamydia trachomatis cytotoxin locus associated with ocular and genital isolates. Infect Immun. 2004;72:7063–72. DOIPubMedGoogle Scholar
- Fine D, Dicker L, Mosure D, Berman S. Increasing chlamydia positivity in women screened in family planning clinics: do we know why? Sex Transm Dis. 2008;35:47–52. DOIPubMedGoogle Scholar
- Dean D, Kandel RP, Adhikari HK, Hessel T. Multiple Chlamydiaceae species in trachoma: implications for disease pathogenesis and control. PLoS Med. 2008;5:e14. DOIPubMedGoogle Scholar
- Somboonna N, Mead S, Liu J, Dean D. Discovering and differentiating new and emerging clonal populations of Chlamydia trachomatis with a novel shotgun cell culture harvest assay. Emerg Infect Dis. 2008;14:445–53. DOIPubMedGoogle Scholar
- McLean CA, Stoner BP, Workowski KA. Treatment of lymphogranuloma venereum. Clin Infect Dis. 2007;44(Suppl 3):S147–52. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 15, Number 9—September 2009
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Deborah Dean, Director, Children’s Global Health Initiative Children's Hospital Oakland Research Institute, 5700 Martin Luther King Jr Way, Oakland, CA 94609, USA;
Top