Volume 17, Number 6—June 2011
Dispatch
Worldwide Distribution of Major Clones of Listeria monocytogenes
Cite This Article
Citation for Media
Abstract
Listeria monocytogenes is worldwide a pathogen, but the geographic distribution of clones remains largely unknown. Genotyping of 300 isolates from the 5 continents and diverse sources showed the existence of few prevalent and globally distributed clones, some of which include previously described epidemic clones. Cosmopolitan distribution indicates the need for genotyping standardization.
Listeria monocytogenes is a foodborne pathogen that can cause listeriosis, a severe invasive infection in humans with a particularly high case-fatality rate. Listeriosis is a major public health concern in all world regions, with an increasing incidence in Europe, especially among elderly persons (1,2).
L. monocytogenes is genetically heterogeneous (3–5). To help epidemiologic investigation and to define clones, i.e., groups of genetically similar isolates descending from a common ancestor, a variety of typing methods have been used, including pulsed-field gel electrophoresis (5,6), single nucleotide polymorphism typing (7), and multiple housekeeping and virulence gene sequencing (8,9). Some clones implicated in multiple outbreaks have been defined as epidemic clones (EC) (3,5,9–11). ECI and ECIV have been described in several countries (3,5), but because of the lack of standardization of genotyping, a definition of clones is not widely accepted, and current knowledge on the global distribution of L. monocytogenes clones is virtually absent. Multilocus sequence typing (MLST) is a reference method for global epidemiology and population biology of bacteria, and its application to L. monocytogenes (12) effectively allows isolate comparisons across laboratories (www.pasteur.fr/mlst). The aim of this study was to investigate the global distribution of L. monocytogenes MLST-defined clones.
Three hundred L. monocytogenes isolates were collected from different sources from 42 countries on 5 continents (Table A1). The isolates derived from 1) the collection of the World Health Organization Collaborating Center for Listeria and 2) the Seeliger Listeria Culture Collection. When available, up to 10 countries per continent were included. Only 1 isolate per documented outbreak was kept, and the isolates from a given country were selected from various sources, years, and serotypes. A total of 117 isolates were from humans, 107 from food, 28 from animals, 32 from the environment and vegetation, and 16 of undocumented origin. The relative proportion of isolates from distinct sources was similar among world regions (Table A1), except that no animal isolate was available from the Western Hemisphere and that the ratio of human to food isolates was lower from this continent.
Each isolate was hemolytic when streaked for isolation on blood agar. Genomic DNA was extracted by using Promega Wizard Genomic DNA purification kit (Promega, Madison, WI, USA). Serotype information was confirmed by PCR serogrouping (13). MLST was performed as described (12). Alleles and sequence types (STs) are publicly available at www.pasteur.fr/mlst. Clonal complexes (CC) were defined as groups of STs differing by only 1 gene from another member of the group (12) and were considered as clones. The θ estimator of the Fst statistic, which measures population differentiation, was determined on the basis of ST frequency by using FSTAT (www2.unil.ch/popgen/softwares/fstat.htm).
Figure A1
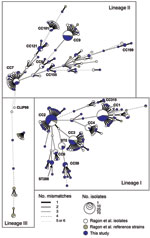
Figure A1. Genetic relationships among 660 Listeria monocytogenes isolates. The graph is a minimum spanning tree based on allelic profiles by using BioNumerics version 6.1 (Applied-Maths, Sint-Martens-Latem, Belgium). The 300 isolates of this...
The 300 isolates represented 111 STs (diversity index 95.4%) grouped into 17 CCs (Figure A1). Phylogenetic analysis of the concatenated genes (not shown) indicated that 199, 98, and 3 isolates belonged to lineages I, II, and III, respectively (12). In lineage I, 3 CCs were highly prevalent: CC1 (47 isolates, serotype 4b), CC2 (64 isolates, 4b,) and CC3 (32 isolates, 1/2b). The remaining isolates of lineage I were of serotype 4b or 1/2b (Table). In lineage II, CC9 (28, all with serotype 1/2c, except one 1/2a isolate) was the most frequent, followed by CC7 (15 1/2a isolates). All other lineage II isolates had serotype 1/2a.
Comparisons of populations from different sources (Table) showed a clear partitioning of genotypic diversity between clinical isolates on the one hand and food or environmental isolates on the other (θ = 0.033 and 0.050, respectively; p<0.0002). Consistent with common knowledge (4,5), and even though recent outbreaks in Canada and Austria/Germany were caused by 1/2a strains, isolates of serotype 4b were, compared with other serotypes, relatively more frequent in human cases than in food. This difference in source distribution was further demonstrated for individual clones because the human/food ratio of both CC1 (2.6) and CC2 (2.8) differed significantly from those of CC3 (0.65) and CC9 (0.5) (χ2 p<0.01 for the 4 comparisons).
A global distribution of L. monocytogenes clones was evident (Figure). Frequent clones were found in many countries (up to 30 countries for CC2; Table A1) and were globally distributed. Remarkably, CC1 and CC2 were predominant in all world regions except northern Africa for CC1 (Figure). CC3 ranked among the 4 most common clones in all regions, whereas CC9 ranked third in Europe and the Western Hemisphere. Altogether, these 4 clones represented 54 (50%) food isolates and 80 (68%) clinical isolates. Our results show that the same few clones account for a large fraction of nonepidemic L. monocytogenes isolates in distant world regions. However, continents and sources were not equally represented in our sample, and larger studies are needed to confirm our hypothesis that the clonal composition is similar across world regions and countries. Consistent with their cosmopolitan distribution, 15 of the 17 clones found herein (except CC199 and CC315, with only 6 and 3 isolates, respectively) included isolates from our previous analysis of 360 isolates, mostly from France (12).
This study provides the first global view of L. monocytogenes clonal diversity. Our results clearly demonstrate the worldwide distribution and high prevalence of a few frequent clones in distinct world regions. In the current debate on the phylogeography of bacterial species (14), major L. monocytogenes clones clearly fit in the “everything is everywhere” group, as do other pathogens in the environment, e.g., Pseudomonas aeruginosa (15). Dispersal by human travel, animal or food trade, wild animal migration, or wind and dust all might contribute to the global diffusion of L. monocytogenes clones. However, finer phylogenetic resolution will possibly subdivide widespread MLST-defined clones into subclades that might exhibit phylogeographic partitioning and will better clarify the rate and patterns of strain dispersal.
Remarkably, some ECs correspond with highly prevalent clones. ECII, described relatively recently (6), and ECIII, involved in outbreaks from a single plant, correspond to 2 clones (CC6 and ST11, respectively [12]), that were rare herein (5 and 0 isolates, respectively), suggesting that both clones experienced particular conditions that favored their diffusion on specific occasions. In contrast, the outbreaks caused by ECI and ECIV, reference strains of which belong to CC1 and CC2, respectively (12), could have been favored by their high prevalence in sources. One important question for future research is whether ECs correspond entirely to MLST-defined clones (i.e., CCs) or whether, on the contrary, they represent a genotypic subset thereof. The cosmopolitan distribution of clones, which protects them against extinction resulting from local disturbances, further highlights the crucial need to standardize L. monocytogenes genotyping to improve global epidemiologic knowledge and monitoring of current emergence trends.
Acknowledgments
We thank all laboratories that have sent isolates to the World Health Organization Collaborating Center for Listeria.
This study was supported by Institut Pasteur (Paris, France) and by the Institut de Veille Sanitaire (Saint-Maurice, France).
References
- Denny J, McLauchlin J. Human Listeria monocytogenes infections in Europe—an opportunity for improved European surveillance. Euro Surveill. 2008;13:pii:8082.
- Goulet V, Hedberg C, Le Monnier A, de Valk H. Increasing incidence of listeriosis in France and other European countries. Emerg Infect Dis. 2008;14:734–40. DOIPubMedGoogle Scholar
- Piffaretti JC, Kressebuch H, Aeschbacher M, Bille J, Bannerman E, Musser JM, Genetic characterization of clones of the bacterium Listeria monocytogenes causing epidemic disease. Proc Natl Acad Sci U S A. 1989;86:3818–22. DOIPubMedGoogle Scholar
- Wiedmann M, Bruce JL, Keating C, Johnson AE, McDonough PL, Batt CA. Ribotypes and virulence gene polymorphisms suggest three distinct Listeria monocytogenes lineages with differences in pathogenic potential. Infect Immun. 1997;65:2707–16.PubMedGoogle Scholar
- Kathariou S. Listeria monocytogenes virulence and pathogenicity, a food safety perspective. J Food Prot. 2002;65:1811–29.PubMedGoogle Scholar
- Evans MR, Swaminathan B, Graves LM, Altermann E, Klaenhammer TR, Fink RC, Genetic markers unique to Listeria monocytogenes serotype 4b differentiate epidemic clone II (hot dog outbreak strains) from other lineages. Appl Environ Microbiol. 2004;70:2383–90. DOIPubMedGoogle Scholar
- Ward TJ, Usgaard T, Evans P. A targeted multilocus genotyping assay for lineage, serogroup, and epidemic clone typing of Listeria monocytogenes. Appl Environ Microbiol. 2010;76:6680–4. DOIPubMedGoogle Scholar
- Lomonaco S, Chen Y, Knabel SJ. Analysis of additional virulence genes and virulence gene regions in Listeria monocytogenes confirms the epidemiologic relevance of multi-virulence-locus sequence typing. J Food Prot. 2008;71:2559–66.PubMedGoogle Scholar
- den Bakker HC, Fortes ED, Wiedmann M. Multilocus sequence typing of outbreak-associated Listeria monocytogenes isolates to identify epidemic clones. Foodborne Pathog Dis. 2010;7:257–65. DOIPubMedGoogle Scholar
- Kathariou S. Foodborne outbreaks of listeriosis and epidemic-associated lineages of Listeria monocytogenes. In: Torrence ME, Isaacson RE, editors. Microbial food safety in animal agriculture. Ames (IA): Iowa State University Press; 2003. p. 243–56.
- Chen Y, Zhang W, Knabel SJ. Multi-virulence-locus sequence typing identifies single nucleotide polymorphisms which differentiate epidemic clones and outbreak strains of Listeria monocytogenes. J Clin Microbiol. 2007;45:835–46. DOIPubMedGoogle Scholar
- Ragon M, Wirth T, Hollandt F, Lavenir R, Lecuit M, Le Monnier A, A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 2008;4:e1000146. DOIPubMedGoogle Scholar
- Doumith M, Buchrieser C, Glaser P, Jacquet C, Martin P. Differentiation of the major Listeria monocytogenes serovars by multiplex PCR. J Clin Microbiol. 2004;42:3819–22. DOIPubMedGoogle Scholar
- Martiny JB, Bohannan BJ, Brown JH, Colwell RK, Fuhrman JA, Green JL, Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol. 2006;4:102–12. DOIPubMedGoogle Scholar
- Pirnay JP, Matthijs S, Colak H, Chablain P, Bilocq F, Van Eldere J, Global Pseudomonas aeruginosa biodiversity as reflected in a Belgian river. Environ Microbiol. 2005;7:969–80. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this study.
2These authors contributed equally to this study.
Table of Contents – Volume 17, Number 6—June 2011
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Sylvain Brisse. Institut Pasteur, Genotyping of Pathogens and Public Health (PF8), 28 Rue du Dr Roux, F-75724 Paris, France
Top