Volume 19, Number 7—July 2013
Letter
Novel Bat-borne Hantavirus, Vietnam
Cite This Article
Citation for Media
To the Editor: Compelling evidence of genetically distinct hantaviruses (family Bunyaviridae) in multiple species of shrews and moles (order Soricomorpha, families Soricidae and Talpidae) across 4 continents (1–7) suggests that soricomorphs, rather than rodents (order Rodentia, families Muridae and Cricetidae), might be the primordial hosts (6,7). Recently, the host range of hantaviruses has been further expanded by the discovery that insectivorous bats (order Chiroptera) also serve as reservoirs (8,9). Conjecturing that Mouyassué virus in the banana pipistrelle (Neoromicia nanus) in Côte d’Ivoire (8) and Magboi virus (MGBV) in the hairy split-faced bat (Nycteris hispida) in Sierra Leone (9) represent a much broader geographic distribution of bat-borne hantaviruses, we analyzed tissues from bats captured in Mongolia and Vietnam.
Total RNA was extracted from 51 lung tissues, collected in RNAlater Stabilization Reagent (QIAGEN, Valencia, CA, USA), from insectivorous bats, representing 7 genera and 12 species, captured in Mongolia and Vietnam. cDNA was then prepared by using PrimeScript II 1st strand cDNA Synthesis Kit (Takara Bio, Otsu, Shiga, Japan) for reverse transcription PCR (RT-PCR), and using oligonucleotide primers previously designed for amplification of soricid- and talpid-borne hantaviruses (1–7).
A novel hantavirus, designated Xuan Son virus (XSV), was detected in 1 of 5 Pomona roundleaf bats (Hipposideros pomona) by using a heminested large (L)–segment primer set (outer: HNL-2111F, 5′-CARTCWACWGTIGGIGCIAGTGG-3′, and HAN-L-R1, 5′-AACCADTCWGTYCCRTCATC-3′; inner: HNL-2111F and HAN-L-R2, 5′-GCRTCRTCWGARTGRTGDGCAA-3′) and a nested small (S)–segment primer set (outer: OSM55F, 5′-TAGTAGTAGACTCC-3′, and XSV-S6R, 5′-AGITCIGGRTCCATRTCRTCICC-3′; inner: Cro-2F, 5′-AGYCCIGTIATGRGWGTIRTYGG-3′, and JJUVS-1233R, 5′-TCACCMAGRTGRAAGTGRTCIAC-3. The bat was captured during July 2012 in Xuan Son National Park, a nature reserve in Thanh Sơn District, Phu Tho Province, ≈100 km west of Hanoi (21°07′26.75′′N, 104°57′29.98′′E).
For confirmation, RNA extraction and RT-PCR were performed independently in a laboratory in which hantaviruses had never been handled. After initial detection, the L-segment sequence was extended by using another primer set (PHL-173F: 5′-GATWAAGCATGAYTGGTCTGA-3′; and TNL-5084R: 5′-GATCCTGAARTACAATGTGCTGG-3′). To calculate the number of virus copies in tissues by real-time RT-PCR, we used a virus-specific primer set (XSV-F: 5′-GTTGCACAGCTTGGTATTGG-3′; and XSV-R: 5′-TTAGCACCCAAACCTCCAAG-3′) and probe (XSV-Probe: 5′-ACAGCTCCTGGCATGGTAAATTCTCC-3′).
Pairwise alignment and comparison (with ClustalW, www.clustal.org) of a 4,582-nt (1,527 aa) region of the RNA-dependent RNA polymerase–encoding L segment indicated sequence similarities of 71.4%–71.5% and 75.9%–78.7% at the nucleotide and amino acid levels, respectively, between XSV and Mouyassué virus and MGBV. Sequence analysis of a 499-nt (166 aa) region of the nucleocapsid-encoding S segment showed that XSV differed by 42.8%–58.3% from representative hantaviruses harbored by rodents and most soricomorphs. XSV sequences were identical in lung, liver, kidney, and spleen; and the highest number of virus copies (7.6 × 101) was in lung tissue, determined by real-time RT-PCR. No additional hantavirus-infected Pomona roundleaf bats were found by RT-PCR that used XSV-specific primers.
Figure
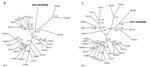
Figure. . Phylogenetic trees, based on 499-nt and 4,582-nt regions of the small (S) and large (L) genomic segments, respectively, of Xuan Son virus (XSV VN1982B4) (GenBank accession nos. S: KC688335, L:...
Phylogenetic analyses was performed with maximum-likelihood and Bayesian methods, and we used the GTR+I+Γ model of evolution, as selected by the hierarchical likelihood-ratio test in MrModeltest version 2.3 and jModelTest version 0.1 (10), partitioned by codon position. Results indicated 4 distinct phylogroups, with XSV sharing a common ancestry with MGBV (Figure). Similar topologies, supported by high bootstrap (>70%) and posterior node (>0.70) probabilities, were consistently derived when various algorithms and different taxa and combinations of taxa were used. Moreover, as we reported previously, the incongruence between some hantaviruses and their reservoir hosts might be indicative of host-switching events (5–7).
The striking sequence divergence of XSV presented considerable challenges for designing suitable primers for RT-PCR and sequencing. Also, sequencing efforts were constrained by the limited availability of tissues and concurrent virus isolation attempts. Consequently, we were unable to obtain the full-length sequence of XSV. Similarly, the inability to detect hantavirus RNA in tissues from other species of bats in this study might be attributed to several factors, including the highly focal nature of hantavirus infection, small sample sizes of bats of any given species, primer mismatches, and suboptimal cycling conditions.
Bats of the genus Hipposideros, family Hipposideridae, are among the most speciose insectivorous bats; ≈70 species are distributed across Africa, Europe, Asia, and Australia. Pomona roundleaf bats are frequently found in or near limestone or sandstone caves. Their colony sizes vary from few to many hundreds of individuals. The vast geographic distribution of the Pomona roundleaf bat throughout Vietnam and in Bangladesh, Cambodia, China, India, Laos, Malaysia, Myanmar, Nepal, and Thailand, provides opportunities to ascertain the genetic diversity and phylogeography of XSV and XSV-related hantaviruses. In this regard, although hantavirus RNA was not detected in archival tissues from bats of ≈20 genera, including several other Hipposideros species (8,9), many more genetically divergent hantavirus species are probably harbored by insectivorous bats. Not all orphan viruses warrant intensive study at the time of their discovery. However, insights into the ecology and transmission dynamics of newfound bat-borne hantaviruses might prepare us to more rapidly diagnose future outbreaks caused by emerging hantaviruses.
Acknowledgments
We thank Hitoshi Suzuki, Shinichiro Kawada, and Kimiyuki Tsuchiya for supporting field investigations and offering helpful suggestions.
This work was supported in part by a grant-in-aid from the Ministry of Health, Labor and Welfare of Japan (Research on Emerging and Re-emerging Infectious Diseases, Health Science Research Grants), the Japan Society for the Promotion of Science (24405045), and the National Foundation for Science and Technology Development of Vietnam (106.11-2012.02).
References
- Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, Novel hantavirus sequences in shrew, Guinea. Emerg Infect Dis. 2007;13:520–2. DOIPubMedGoogle Scholar
- Arai S, Song J-W, Sumibcay L, Bennett SN, Nerurkar VR, Parmenter C, Hantavirus in northern short-tailed shrew, United States. Emerg Infect Dis. 2007;13:1420–3. DOIPubMedGoogle Scholar
- Song J-W, Kang HJ, Song KJ, Truong TT, Bennett SN, Arai S, Newfound hantavirus in Chinese mole shrew, Vietnam. Emerg Infect Dis. 2007;13:1784–7. DOIPubMedGoogle Scholar
- Song J-W, Kang HJ, Gu SH, Moon SS, Bennett SN, Song KJ, Characterization of Imjin virus, a newly isolated hantavirus from the Ussuri white-toothed shrew (Crocidura lasiura). J Virol. 2009;83:6184–91. DOIPubMedGoogle Scholar
- Arai S, Ohdachi SD, Asakawa M, Kang HJ, Mocz G, Arikawa J, Molecular phylogeny of a newfound hantavirus in the Japanese shrew mole (Urotrichus talpoides). Proc Natl Acad Sci U S A. 2008;105:16296–301. DOIPubMedGoogle Scholar
- Kang HJ, Bennett SN, Sumibcay L, Arai S, Hope AG, Mocz G, Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea). PLoS ONE. 2009;4:e6149. DOIPubMedGoogle Scholar
- Kang HJ, Bennett SN, Hope AG, Cook JA, Yanagihara R. Shared ancestry between a mole-borne hantavirus and hantaviruses harbored by cricetid rodents. J Virol. 2011;85:7496–503. DOIPubMedGoogle Scholar
- Sumibcay L, Kadjo B, Gu SH, Kang HJ, Lim BK, Cook JA, Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Côte d’Ivoire. Virol J. 2012;9:34. DOIPubMedGoogle Scholar
- Weiss S, Witkowski PT, Auste B, Nowak K, Weber N, Fahr J, Hantavirus in bat, Sierra Leone. Emerg Infect Dis. 2012;18:159–61. DOIPubMedGoogle Scholar
- Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–6. DOIPubMedGoogle Scholar
Figure
Cite This ArticleRelated Links
Table of Contents – Volume 19, Number 7—July 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Satoru Arai, Infectious Disease Surveillance Center, National Institute of Infectious Diseases, Toyama 1-23-1, Shinjuku, Tokyo 162-8640, Japan
Top