Volume 2, Number 4—October 1996
Synopsis
Molecular Mechanisms of Bacterial Virulence: Type III Secretion and Pathogenicity Islands
Secretion Systems in Bacteria
Type III Systems Secrete Effector Proteins Upon Contacting Host Cells
How Pathogens Use Type III Secretion Systems in the Host
The Nuts and Bolts of Type III Secretion
Where Do Type III Secretion Systems Come From?
History and Definition of "Pathogenicity Islands"
Pathogenicity Islands Contain Virulence Genes and Regulatory Elements
Pathogenicity Islands Can Exist in Various Structural Types and Numbers Within a Bacterium
Possible Origins of Pathogenicity Island DNA and Mechanisms of Transfer and Insertion
Relative Advantages of Instability and Stability
Foreign DNA Is a Significant Determinant in Recently Emerged Pathogens
Benefiting From Information About Type III Secretion and Pathogenicity Islands
Cite This Article
Abstract
Recently, two novel but widespread themes have emerged in the field of bacterial virulence: type III secretion systems and pathogenicity islands. Type III secretion systems, which are found in various gram-negative organisms, are specialized for the export of virulence factors delivered directly to host cells. These factors subvert normal host cell functions in ways that seem beneficial to invading bacteria. The genes encoding several type III secretion systems reside on pathogenicity islands, which are inserted DNA segments within the chromosome that confer upon the host bacterium a variety of virulence traits, such as the ability to acquire iron and to adhere to or enter host cells. Many of these segments of DNA appear to have been acquired in a single step from a foreign source. The ability to obtain complex virulence traits in one genetic event, rather than by undergoing natural selection for many generations, provides a mechanism for sudden radical changes in bacterial-host interactions. Type III secretion systems and pathogenicity islands must have played critical roles in the evolution of known pathogens and are likely to lead to the emergence of novel infectious diseases in the future.
In the past decade, there has been an explosion of new information about bacterial pathogens as researchers have begun to examine the molecular and genetic bases of microbial pathogenicity. Because microbes invade many niches in humans and cause a wide variety of syndromes, it initially appeared that each disease might be created by a distinct molecular mechanism. However, the spectrum of methods is not as broad as first imagined; rather, bacteria exploit a number of common molecular tools to achieve a range of goals (1). Among these tools are pathogenicity islands, which enable bacteria to gain complex virulence traits in one step, and type III secretion systems, which provide a means for bacteria to target virulence factors directly at host cells. These factors then tamper with host cell functions to the pathogens' benefit.
Early in the search for virulence genes, researchers discovered that many of these genes resided on plasmids or phages; however, it was also clear that these genes did not produce all of the physiologic changes induced in host cells by various pathogens (2). Thus, researchers searched the chromosome. Surprisingly, as when found on plasmids, virulence genes often clustered in functionally related groups. Furthermore, these groups often appeared to have been acquired from another organism, as features of their DNA sequence differed from the bulk of the genome. These observations gave rise to the concept of pathogenicity islands—discrete segments of DNA that encode virulence traits and often appear to have a foreign origin (3,4).
Researchers found that a particular set of virulence genes appeared several times on both plasmids and pathogenicity islands (5-8). These genes were discovered in both plant and animal pathogens and were homologous to genes encoded on a virulence plasmid of pathogenic Yersinia spp. (Table 1) (9-12). The Yersinia proteins are the components of a novel secretion system (13), called type III (14). This machinery propels effector molecules toward host cells where they alter host physiology (15,16). The homology suggested that many divergent bacterial pathogens had acquired a similar system from a common source. Pathogens use the type III system to secrete different effector molecules that influence host cells in a variety of ways (16-19).
Figure 1
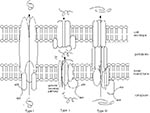
Figure 1. Schematic diagram of type I, type II, and type III secretion systems. All systems use the energy of ATP hydrolysis to drive secretion. Type I and type III secrete proteins across...
Secreted or surface-exposed bacterial proteins have long been known to play central roles in bacterial-host interactions. In gram-negative bacteria, these proteins must pass through two membranes: the inner membrane, which surrounds the cytoplasm, and the outer envelope, which encloses the periplasm and acts as a barrier to the environment (Figure 1). The general secretory pathway transports proteins to the periplasm. Before the Yersinia secretion system was identified, two other specialized secretion systems, type I and type II, were known to transport molecules to the cell surface (14,20,21). Proteins secreted by the Type I system cross directly from the cytoplasm to the cell surface, bypassing the general secretory pathway completely (Figure 1). Type II-secreted proteins use the general secretory pathway to reach the periplasm and then traverse the outer membrane through distinct channel proteins. Both type I and type II systems secrete proteins involved in various functions, including pathogenesis. For example, -hemolysin of E. coli uses a type I system and bundle-forming pili of enteropathogenic E. coli (EPEC) and enterotoxigenic E. coli (ETEC) use type II systems for export.
In the past 5 years, the highly conserved, multicomponent type III secretion system has been found in many gram-negative bacteria that cause disease in animals and plants (8). This secretion system is responsible for transporting effector molecules directly from the cytoplasm to the cell surface, where they interact with mammalian cells and modify host cell proteins (13). This one-step secretion process is reminiscent of the mechanism used by type I systems (Figure 1). The genes that encode many components of type III systems are homologous to those that encode flagellar export machinery in both gram-negative and gram-positive bacteria (Table 1) (22-24). Indeed, these two systems share many structural and functional features. The differences reside at the outer membrane. Flagellar components pass through an outer ring structure that is part of the flagellum itself (23), whereas pathogenic effector molecules traverse the outer membrane through a channel protein that is homologous to those used in type II secretion systems (20,25). While our discussion of type III secretion systems will focus primarily on those used by human pathogens, many characteristics are common among systems found in plant pathogens and bacteria that produce flagella.
In contrast to the secretion process in type I and type II systems, type III secretion is triggered when a pathogen comes in close contact with host cells (18,19,26,27), and hence, has been called contact-dependent secretion (28). Temperature, growth phase, and salt conditions are environmental cues known to induce synthesis of the secretion apparatus and effector molecules in various pathogens (29-31). When the pathogen comes into close contact with tissue culture cells, effector molecules move to the external surface of the bacterium, sometimes forming appendages suggestive of flagellae (18). In some cases, the bacterium binds to the host cells and these molecules are delivered into the host cell (32). The effector molecules cause changes in host cell function, which facilitate the pathogen's ability to survive and replicate (15-17,33).
The best studied bacterial pathogens that use type III secretion are Yersinia pestis, which causes plague, and a number of enteropathogens. Although these various enteropathogens (Yersinia spp., Salmonella spp., Shigella spp., and EPEC) cause diarrhea and, in some cases, systemic disease, they produce distinct syndromes because their secreted proteins target different host cells and molecules (Table 1) (34).
Yersinia spp. use their effector molecules to destroy key functions of immune cells and render them innocuous (35). When these bacteria bind to tissue culture cells, approximately 10 different effector molecules are secreted (13) and at least three are injected into cells (27,36-38). Two of these injected molecules, YopE and YopH, modify macrophage proteins and destroy the cells' abilities to engulf and kill bacteria (16,39). During the course of disease, immune cells are presumably neutralized by these effector molecules, which enables Yersinia spp. to flourish in the reticuloendothelial environment.
While effector molecules in Yersinia destroy normal cellular functions, those from Shigella spp. and from one of the Salmonella spp. type III secretion systems, encoded by genes located in SPI I (Table 2), stimulate cells to perform functions in addition to those in their usual repertoires. Studies in epithelial tissue culture systems show that these bacteria induce their own entry into normally nonphagocytic cells by using effector molecules secreted by their type III systems (28,40). During the course of disease, Shigella spp. enter and replicate in the mucosal epithelial cells of the large intestine, while Salmonella spp. gain entry into the peritoneal cavity by passing through the epitheloid-like M cells in the small intestine (41). In the murine model for typhoid fever, S. typhimurium that are defective in this secretion system are attenuated for infection when administered orally, but not intraperitoneally (42). Presumably this attenuation is a consequence of reduced entry into the M cells in the small intestine, a barrier that is bypassed by intraperitoneal delivery.
Although neither its effector molecules nor target host cells have been identified, a second type III secretion system, encoded by genes located in SPI II (Table 2), has been described in S. typhimurium, on the basis of sequence homology (43). The genes in SPI II, in contrast to those in SPI I, are required for systemic disease regardless of the route of infection (43,44). Presumably the factors encoded in SPI II act after the bacteria have crossed the epithelial barrier of the small intestine.
Although the functions and sites of action of the secretion systems differ among these enteric pathogens, effector molecules from one system can be secreted by other systems, provided the appropriate chaperones are present. Such heterologously expressed effector molecules can induce the same cellular response as when expressed from their native systems. For instance, an effector molecule from Yersinia that causes actin depolymerization has the same effect on tissue culture cells when secreted from Salmonella (45). Likewise, proteins of Shigella and Salmonella involved in bacterial uptake into cultured epithelial cells are functionally interchangeable (46).
The observation that Salmonella spp. have two contact-dependent systems that function at distinct stages to cause disease raises several interesting questions. Why do Salmonella spp. need different type III secretion apparatuses when it is clear that effector molecules can be secreted from heterologous systems? Could one suffice if the two sets of effector molecules were expressed at appropriate times during the course of infection? Alternatively, do these two sets of effector molecules need to be delivered to different target cells in a specific manner, which is only possible with distinctly customized machinery? Answers to these questions will illuminate issues about both the course of salmonellosis and the basic mechanics of the secretion apparatus.
The type III secretion apparatus in Yersinia spp. has been the most intensively investigated. However, this work has been done in three Yersinia spp; thus several proteins have been shown to be essential for effector molecule secretion in one species but have not yet been examined in others. Our analysis of these studies assumes that proteins essential in one will play a similar role in all (Table 1).
One essential feature of any secretion system is that energy must be provided to move molecules through the membrane (14). Only one protein in the system, YscN, has been shown to hydrolyze ATP and thus is a likely candidate for generating energy to drive secretion (47). YscN is predicted to be a cytoplasmic protein, closely associated with the inner membrane.
Several proteins essential to secretion including LcrD, YscD, R, S, T, and U, are known or predicted to reside in the inner membrane (10,12,48-50). At the outer membrane, only one protein, YscC (48), and two lipoproteins, YscJ and VirG (51,52), appear essential for proper secretion. The roles and subcellular locations are not known for several more essential proteins, YscE, F, G, I, K, and L (9,48,51). How all of these proteins interact with one another to form the secretion apparatus is not yet understood. It is clear, however, that correct assembly of the apparatus is required not only for secretion, but also for normal synthesis of effector molecules (47,48). If one component of the export machinery is missing, production of the effector molecules is altered. This feedback regulation also occurs in systems that produce flagella (53). How it works in Yersinia is under investigation; thus far, only one protein, LcrQ, has been implicated (54).
Two proteins, YopB and YopD, are loosely associated with the outer membrane (55) and are crucial for efficient delivery of effector molecules into target cells. These two proteins use the type III secretion system to reach the bacterial cell surface. Without YopB, which has homology to pore-forming toxins (55), and YopD, effector molecules are secreted but not efficiently internalized by host cells; thus, their activities on host cells are severely abrogated (27,36). Presumably, YopB and YopD form a mammalian cell through which effector molecules pass.
Several proteins, called chaperones, play critical roles in secretion by binding to effector molecules in the bacterial cytoplasm. Chaperones have several proposed functions (56-58). Chaperone binding may stabilize and prevent proteins from folding into conformations that are impossible to secrete. Alternatively, as has been shown for Shigella, they may prevent effector molecules from improperly associating with one another before secretion (58). Lastly, chaperones may deliver molecules to the secretion apparatus.
In addition to the feedback regulation mentioned above, the synthesis of and of and secretion from the Yersinia type III system is regulated by two networks that respond to environmental cues (29,35). A temperature-sensing network induces synthesis of the apparatus at 37°C and includes VirF and YmoA (35). A host cell-sensing network increases both synthesis of and secretion from the type III system when Yersinia binds to target cells. This regulatory system is called the low-calcium response network—low calcium presumably mimics some signal generated by cell contact—and includes YopN, LcrG, and LcrQ (32,54,59,60). YopN localizes to the outer membrane, where it senses cell contact and transduces this signal to the cytoplasm by an unknown mechanism (60). The role of LcrG has not yet been elucidated. LcrQ functions as a repressor of the Yops. When Yersinia comes into close contact with host cells, LcrQ is secreted from the cytoplasm through the type III secretion system. This lowers the intracellular concentration of LcrQ and results in an increase in synthesis and secretion of the Yops (32). Flagella synthesis is controlled in a similar manner (61).
Many of the structural components of the Yersinia system have homologues in Shigella, Salmonella, and EPEC (Table 1). A comparison of proteins found in each system shows that certain core structural components are present in all type III apparatuses, whereas others may exist in only one or a subset. These differences may be due to particular functions of each system.
Several studies have examined whether structural components from different bacteria are interchangable. In general, core constituents from Shigella and SPI I of Salmonella, which both facilitate bacterial uptake by epithelial cells, are interchangable with one another, but not with those in the Yersinia system (7,62). These results may be due to the observation that factors from Shigella and SPI I of Salmonella are predicted by sequence homology to be more structurally similar to each other than to those in Yersinia (7). Alternatively, some of the regulatory cues for secretion and assembly may be different for Yersinia than for Salmonella and Shigella.
Most proteins in the type III secretion systems, including effector proteins, regulatory proteins, structural proteins, and chaperones (Table 1), are encoded by genes that belong to several large operons, which are clustered together (7,9,12,63). These operons are on plasmids in some species, and on the chromosome in others (Table 1). In some cases, such as in Shigella and SPI I of Salmonella, the order of the genes within operons and the arrangement of the operons with respect to each other are conserved (7). These observations suggest that type III systems were inherited en masse, and likewise, could be transmitted to other bacteria en masse. One can speculate that the acquisition of a type III secretion system could allow a bacterium to adapt to different environments or hosts. For instance, a new pathogen could perhaps arise if a skin-commensal bacterium were to acquire the means to penetrate and survive in the skin-associated lymphoid tissue by obtaining a type III secretion system from an enteropathogen.
It seems plausible that the original type III secretion sysem for virulence factors evolved from those for flagellar assembly (22,53). The bacterial flagellum exists in a wide range of eubacteria and some archaebacteria, which indicates that it probably emerged well before gram-negative bacteria, the hosts of the type III virulence factor secretion systems identified thus far.
Attempts to establish any of the known type III secretion systems as the progenitor have been fruitless. On the basis of degrees of homology among different type III systems and well-established evolutionary relationships between the bacteria, each organism can be ruled out as the source (64). For example, Shigella emerged from E. coli after Salmonella and E. coli diverged from a common ancestor; thus, Shigella cannot have provided the type III systems conserved in the Salmonella spp. Conversely, as Shigella type III apparatus sequences have a G+C content well below that of Salmonella and of the bulk of the Shigella chromosome, Salmonella could not have been the source of the Shigella genes (Table 2). Thus, the ability to secrete effector molecules by this mechanism seems to have been introduced independently into each of these bacteria.
Examination of homologous genes in the epithelial cell invasion loci of Salmonella and Shigella shows that some are highly conserved, while others display much lower levels of homology (65). Li and colleagues have found a relationship between evolutionary rate of change and subcellular location: genes encoding several secreted proteins are hypervariable in relation to genes encoding several proteins located in the bacterial inner membrane (65). In principle, hypervariability could reflect antigenic variation or adaptations to diverse host environments; however, neither of these explanations appears to pertain to the particular proteins examined (28,65).
Type III systems sometimes provide much of what distinguishes particular organisms from closely related, often nonpathogenic, species. As described above, many operons encoding type III secretion machinery are clustered. DNA sequence analysis has shown that these loci are often distinguishable from the bulk of the genomic DNA. The loci that are chromosomally located represent "pathogenicity islands" (66).
The phrase "pathogenicity island" was first used to describe two large, unstable pieces of chromosomal DNA, unique to uropathogenic E. coli, that encode a number of genes required for virulence (3,4). Since its conception, the term has evolved to include regions of expression chromosomal DNA essential for pathogenicity that do not appear to "belong" (Table 2). Not all pathogenicity islands are genetically unstable, but each one shows an indication of foreign origin. These pieces of DNA are often missing in closely related, nonvirulent bacteria. Many pathogenicity islands differ from the bulk of the genome in G+C content and codon usage, and their borders are often marked by repeated sequences or insertion elements, which suggests that some kind of recombination event delivered them to the chromosome. Several encode multiple proteins that collaborate to confer a single, complex virulence property to the bacterial host.
The definition of pathogenicity islands includes chromosomal location. As such, the plasmid-borne type III gene clusters of Yersinia and Shigella do not qualify (Table 1). This seems somewhat arbitrary. Indeed, phages and a number of plasmids can easily insert into and excise from the chromosome. Similarly, many transposable elements replicate and function equally as well in the chromosome as on an extrachromosomal element. It seems to us that a block of apparently foreign genes found uniquely in pathogenic members of a genus and required for virulence is a more useful and relevent defining feature of a pathogenicity island than location. Thus, it makes sense to include the loci encoding type III secretion systems, regardless of whether they reside on a plasmid or chromosome. In the discussion below, however, we adhere to the established definition that includes chromosomal location.
Pathogenicity islands also contain virulence genes other than those encoding type III secretion systems; a common theme appears to be inclusion of genes for secreted or cell surface-localized proteins such as hemolysins, fimbriae, and hemin-binding factors (Table 2). In fact, the similarities between pathogenicity islands extend further: examination of the large ones shows that many also contain genes that encode a secretion system and environmental sensors. They also can include proteins that regulate expression of genes that lie outside the pathogenicity island. For example, pathogenicity island II (Pai II) of uropathogenic E. coli contains genes that encode transcriptional activators of S-fimbrial genes that reside at a chromosomal locus remote from either of the known pathogenicity islands in this species (67).
A single bacterial strain can harbor more than one pathogenicity island. Salmonella contains at least five: the gene clusters encoding the two type III secretion systems described above, sifA (see below), and two groups of genes that are activated by the two-component regulator, PhoP/PhoQ. These loci vary in size and complexity and reside at distinct chromosomal locations (43,68,69, and S. Miller, pers. comm.).
Pathogenicity islands themselves can be composed of distinct segments. For example, an unstable 102-kb region of DNA that encodes several traits important for virulence of Y. pestis appears to consist of several regions (70-72). One contains the hemin storage genes and has a G+C content similar to that of the bulk of the chromosome (R. Perry, pers. comm.); the other contains genes encoding the Yersiniabactin receptor and iron-regulated proteins and has a significantly higher G+C content (R. Perry, pers. comm) (73). Although the 102 kb region often deletes entirely, the two regions can also act independently. In some strains the chromosomal region containing the hemin storage genes spontaneously deletes from the chromosome at a significant frequency, while the Yersiniabactin receptor/iron-regulated protein region appears stable (72). Furthermore, only the Yersiniabactin receptor/iron-regulated segment is present in Y. enterocolitica (70).
Even more complex pathogenicity islands are harbored by strains of Helicobacter pylori, the causative agent of gastritis and peptic ulcer disease in humans. Strains of H. pylori have been divided into two classes: type I strains express the cytotoxin-associated gene A (CagA) antigen and induce secretion of the neutrophil attractant IL-8 by epithelial cells in vitro, while type II strains lack both of these properties. Patients with duodenitis, duodenal ulcers, and gastric tumors are most often infected by type I strains. Likewise, type I strains are more likely than type II strains to cause gastric injuries in murine model systems. Analysis of the chromosomal region that contains the cagA gene has shown that it is a pathogenicity island of approximately 40 kb of DNA, missing in type II strains, and that mutations in this region abolish IL-8 induction in gastric epithelial cell lines (Censini, S et al. A pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated factors. 1996; submitted for publication).
Different type I strains display considerable heterogeneity in the cag region (Censini, S et al. A pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated factors. 1996; submitted for publication). In some isolates, the cag region is interrupted by one or more insertion sequences. In a small number of strains, there is an additional 20-kb sequence that is also present in type II strains. Partial deletions of the cag region have been detected as well. Thus, the cag pathogenicity island appears to be undergoing dynamic changes in natural Helicobacter populations. Further study of the cag region may elucidate details of pathogenicity island acquisition and help correlate regions of the pathogenicity island with disease symptoms in the murine model system.
Since its establishment, the definition of pathogenicity islands has evolved to include genetic regions that are neither large nor complex; single genes of apparently foreign origin can also be inserted into chromosomal DNA. S. typhimurium has recently been shown to contain such a gene, called sifA, which is required for formation of distinctive structures associated with Salmonella-containing vacuoles within epithelial cells and contributes to pathogenicity in the murine typhoid fever model system (Table 2) (69).
While the sources of pathogenicity islands are unknown, their presence in a wide variety of organisms (Table 2) indicates that bacteria can acquire DNA despite multiple barriers to chromosomal gene transfer between species. The existence of "foreign" genomic DNA is particularly intriguing as sequence divergence is a major limitation to such transfers because it severely limits the potential for homologous recombination (74).
Although the identity of the vectors that transport pathogenicity islands from donor to recipient organisms is unknown, any number of mobile genetic elements are candidates. Clear evidence showing an extrachromosomal stage of a pathogenicity island is lacking; however, it is intriguing that the G+C contents of the Helicobacter pathogenicity island and plasmid are similar to each other and distinct from the chromosome (Censini, S et al. A pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated factors. 1996; submitted for publication). Phages, plasmids, transposons, integrons, and even free DNA carry genes from one organism to another (74). Indeed, many phages and plasmids contain virulence genes, and often these loci seem alien to the bacterial species in which they reside (75). Furthermore, the animal host environment may be particularly conducive to DNA transfer events. For example, the phage that encodes cholera toxin infects V. cholerae more efficiently within the gastrointestinal tract of a mammalian host than under laboratory conditions (76).
Pathogenicity islands insert into the chromosome by an unknown mechanism; however, the existence of insertion elements and repeated DNA motifs at the boundaries of several pathogenicity islands suggest that recombination events are involved. Recombination has recently been shown to be the major factor governing the divergence of a group of E. coli strains and is a significant driving force for evolution (74,77). Although the genetic material comprising pathogenicity islands may be introduced into a new host organism in a single step, the events that generate known pathogenicity islands are unlikely to be simple insertions, because DNA rearrangements and alterations are common in the flanking chromosomal regions.
The identification of several tRNA genes as insertion sites for pathogenicity islands is also notable, although the significance of this remains obscure (Table 2). tRNA genes serve as integration sites for a variety of prokaryotic genetic elements, including several phages and transmissible plasmids (78-80). Perhaps the conserved portion of tRNA genes is a useful landmark for mobile genetic elements that inhabit a variety of prokaryotic hosts; in addition, the regions of dyad symmetry characteristic of all tRNA genes could serve as binding sites for enzymes involved in recombination.
Some pathogenicity islands can excise from the chromosome and are apparently lost from the host bacterium (Table 2) (3,4,70). Such instability may provide an adaptive advantage. Virulence properties may be dispensable at certain stages of infection, and the coordinated loss of these characteristics could be beneficial to the bacterium. Indeed, expression of particular genes at inappropriate times can be detrimental to bacterial pathogens (81). Natural selection of strains with deleted virulence regions can occur in specific environments: diabetic patients are more susceptible to uropathogenic E. coli strains not exhibiting virulence phenotypes (4).
On the other hand, particular virulence traits could provide a continual adaptive advantage, resulting in stable pathogenicity islands. The "foreign nature" of pathogenicity islands may reflect this benefit; foreign DNA may be actively maintained in the population because of its limited ability to recombine with related organisms (75). It is not clear whether stable islands exist because of a divergence of sequences at the borders (for example, repeated elements that are no longer recognizable as such), an integration mechanism completely different from that of the unstable islands, or because of a lack of excision machinery.
Deletion of pathogenicity islands can affect gene expression by altering the chromosomal site of insertion and by removing the genes contained in the island. The locus of enterocyte effacement (LEE) comprising the type III secretion apparatus in EPEC and pathogenicity island I (Pai I) of uropathogenic E. coli both insert at the selenocysteine tRNA (selC) gene (82,83). The presence of Pai I does not interfere with selC expression. However, excision from the chromosome appears to occur by a recombination event between the repeated sequences in selC and the distal end of Pai I. This recombination event results in deletion of part of the tRNA gene and inhibits anaerobic growth due to the cell's inability to produce formate dehydrogenase, which contains selenocysteine (84). Similarly, in Pai II deletion strains, the leuX tRNA gene at the insertion site is disrupted, which interferes with its ability to act as a global regulator of several virulence factors that lie outside the pathogenicity island (84).
Bacteria may be able to have the best of both the stable and the unstable worlds. The phenotypic loss of the enteroinvasive E. coli and Shigella flexneri virulence plasmids is sometimes due to plasmid insertion into a specific site on the chromosome (85,86). After integration, excision also can be detected; strains containing precisely excised plasmids regain virulence, while those with imprecisely excised plasmids remain noninvasive. Several of the Y. pestis plasmids exhibit similar behavior (87-89). Integration, which simultaneously maintains these plasmids in the bacterial genome while downregulating their genes, may represent a sophisticated adaptation to the requirements of different environments or may represent? stages in the bacterial life cycle. Furthermore, integration and excision remind us that strict definitions of "chromosomal" versus "plasmid-borne" do not always reflect biological reality.
Horizontal gene transfer has been invoked to explain the origin of enterohemorrhagic E. coli (EHEC), which causes hemorrhagic colitis and hemolytic uremic syndrome (90). Like EPEC, EHEC induces striking morphologic changes—called attaching and effacing (AE) lesions—in host cells of the small intestine; however, unlike EPEC, EHEC contains Shiga-like toxins. After analyzing the genetic relationships between many E. coli strains, Whittam and colleagues proposed that EHEC arose from an EPEC-like progenitor strain, which then acquired the prophage-encoded Shiga-like toxins, thus becoming a new pathogen that expresses both sets of traits (90).
The new epidemic Vibrio cholerae O139 strain may have emerged after acquisition of a pathogenicity island (91). Although it appears that V. cholerae O139 arose from a strain of the same serotype (O1) that is causing the ongoing cholera pandemic (O1 El Tor) (92), V. cholerae O139 contains an additional piece of DNA that replaces part of the O antigen gene cluster of O1 strains (93). The inserted DNA contains open reading frames homologous to proteins involved in capsule and O antigen synthesis, two factors that distinguish O139 and O1 El Tor, and are thought to mediate activities important for pathogenesis and evasion of immunity.
Pathogenic bacteria continue to exhibit impressive genetic flexibility and exchange and use these abilities to adapt to varied types of lifestyles within host organisms. It should be possible to use the information from studies of pathogenicity islands and type III secretion systems in the ongoing characterization of bacterial infections. When a novel pathogen is isolated, it may be worthwhile to identify chromosomal regions specific to it by comparing the gross genomic structure with that of related organisms, which may provide a shortcut to the identification of virulence genes. Likewise, simple molecular techniques can determine whether bacteria contain type III secretion systems, because genes encoding particular components are highly conserved; perhaps this procedure should be part of our standard investigative arsenal as well.
Our knowledge of type III secretion systems may yield therapeutic benefits. The contact-dependent systems appear to reside in pathogenic and not in commensal bacteria. If this observation reflects a general truth, antibiotics that target type III systems may specifically attack intruding bacteria and spare the normal flora; therefore, these antibiotics might produce minimal side effects. In addition, type III secretion systems will provide new targets for therapeutic drugs that might not kill the bacterium but would inhibit the disease process. We also may be able to exploit this secretion system, by using appropriately attenuated bacteria, to prime immunity. Chimeric proteins—fusions between effector and other proteins—can be secreted in large quantities by the type III secretion machinery and be internalized by host cells; furthermore, these proteins can elicit an antibody response in mice (38,94).
The virulence traits of pathogenic microorganisms at the genetic and molecular level remind us that bacterial pathogenicity does not arise by slow adaptive evolution but by "quantum leaps" (95); therefore, microbes can acquire complete systems that radically expand their capabilities to exploit and flourish in different host environments. History teaches us that infectious diseases may change in severity and form, but they will not simply disappear. Microrganisms are, after all, survivors, and there is no escaping our destiny—to be consumed by them in one way or another. However, the more we learn about the microbial tactics of survival, the longer we forestall this destiny.
Joan Mecsas received her Ph.D. at the University of Wisconsin-Madison. She has been a post-doctoral fellow in Dr. Stanley Falkow's laboratory at Stanford University for the past two-and-a-half years. Her work is supported by a Damon Runyon-Walter Winchell Cancer Research Fund postdoctoral fellowship (DRG#1277).
Evelyn J. Strauss received her Ph.D. at the University of California-San Francisco. She has been a post-doctoral fellow in Dr. Stanley Falkow's laboratory at Stanford University for the past 3 years. Her work is supported by an American Cancer Society postdoctoral fellowship (Grant #PF-4120).
Acknowledgment
We thank A. Covacci, D. Frank, R. MacNab, S. Miller, R. Milkman, K. Nelson, R. Perry, K. Rudd, C. Stephens, S. Straley, M. Waldor, R. Welch, and T. Whittam for sharing unpublished data and manuscripts; R. Perry and R. Welch for patient explanations and helpful discussions; S. Fisher, D. Gunn, C. Lee, T. McDaniel, S. Mel, K. Ottemann, B. Raupach, J. Shea, S. Straley, and C. Stephens for thoughtful and incisive comments on the manuscript, and Stanley Falkow for his critical input on the manuscript, the fanciful and poignant concluding paragraph, a stimulating and exciting atmosphere in which to learn and think about bacterial pathogenesis, and his support and mentorship. We apologize to the many researchers whose work we did not cite.
References
- Finlay BB, Falkow S. Common themes in microbial pathogenicity. Microbiol Rev. 1989;53:210–30.PubMedGoogle Scholar
- Hayes W. The Genetics of Bacteria and their Virues. In: 2nd ed. 1968, New York: John Wiley & Sons Inc.
- Knapp S, Hacker J, Jarchau T,Goebel W. Large, unstable inserts in the chromosome affect virulence properties of uropathogenic Escherichia coli O6 strain 536. J Bacteriol. 1986;168:22–30.PubMedGoogle Scholar
- Hacker J, Bender L, Ott M, Wingender J, Lund B, Marre R, Deletions of chromosomal regions coding for fimbriae and hemolysins occur in vitro and in vivo in various extraintestinal Escherichia coli isolates. Microb Pathog. 1990;8:2213–25. DOIGoogle Scholar
- Fenselau S, Balbo I, Bonus U. Determinants of pathogenicity in Xanthomonas campestris pv. vesicatoria are related to proteins involved in secretion in bacteria pathogens of animals. Mol Plant Microbe Interact. 1992;5:390–6.PubMedGoogle Scholar
- Sasakawa C, Komatsu K, Tobe T, Fukuda I, Suzuki T, Yoshikawa M. Eight genes in region 5 that form an operon are essential for invasion of epithelial cells by Shigella flexneri. J Bacteriol. 1993;175:2334–46.PubMedGoogle Scholar
- Groisman EA, Ochman H. Cognate gene clusters govern invasion of host epithelial cells by Salmonella typhimurium and Shigella flexneri. EMBO J. 1993;12:3779–87.PubMedGoogle Scholar
- van Gijsegem F, Gough C, Zischek C, Niqueux E, Arlat M, Genin S, . The hrp gene locus of Pseudomonas solanacearum, which controls the production of type III secretion system, encodes eight proteins related to components of bacterial flagellar biogenesis complex. Mol Microbiol. 1995;15:1095–114. DOIPubMedGoogle Scholar
- Michiels T, Vanooteghem J-C, de Rouvroit C, China B, Gustin A, Boudry P, . Analysis of virC, an operon involved in secretion of Yop proteins by Yersinia enterocolitica. J Bacteriol. 1991;173:4994–5009.PubMedGoogle Scholar
- Plano GV, Barve SS, Straley SC. LcrD, a membrane-bound regulator of the Yersinia pestis low-calcium response. J Bacteriol. 1991;173:7293–303.PubMedGoogle Scholar
- Haddix PL, Straley SC. Structure and regulation of the c Yersinia pestis yscBCDEF operon. J Bacteriol. 1992;174:4820–8.PubMedGoogle Scholar
- Bergman T, Erickson K, Galyov E, Persson C, Wolf-Watz H. The lcrB (yscN/U) gene cluster of Yersinia pseudotuberculosis is involved in Yop secretion and shows high homology to the spa gene clusters of Shigella flexeri and Salmonella typhimurium. J Bacteriol. 1994;176:2619–26.PubMedGoogle Scholar
- Michiels T, Wattiau P, Brasseur R, Ruysschaert J-M,Cornelis GR. Secretion of Yop proteins by Yersiniae. Infect Immun. 1990;58:2840–9.PubMedGoogle Scholar
- Salmond GPC, Reeves PJ. Membrane traffic wardens and protein secretion in Gram-negative bacteria. Trends Biochem Sci. 1993;18:7–12. DOIPubMedGoogle Scholar
- Rosqvist R, Forsberg Å, Wolf-Watz H. Intracellular targetting of the Yersinia YopE cytotoxin in mammalian cells induces actin microfilament disruption. Infect Immun. 1991;59:4562–9.PubMedGoogle Scholar
- Rosqvist R, Forsberg Å, Rimpiläinen M, Bergman T, Wolf-Watz H. The cytotoxic protein YopE of Yersinia obstructs the primary host defense. Mol Microbiol. 1990;4:657–67. DOIPubMedGoogle Scholar
- Ménard R, Prévost M-CGP, Sansonetti P, Dehio C. The secreted Ipa complex of Shigella flexneri promotes entry into mammalian cells. Proc Natl Acad Sci U S A. 1996;93:1254–8. DOIPubMedGoogle Scholar
- Ginocchio CC, Olmsted SB, Wells CL, Galán JE. Contact with epithelial cells induces the formation of surface appendages on Salmonella typhimurium. Cell. 1994;76:717–24. DOIPubMedGoogle Scholar
- Watarai M, Tobe T, Yoshikawa M, Sasakawa C. Contact of Shigella with host cells triggers release of Ipa invasins and is an essential function of invasiveness. EMBO J. 1995;14:2461–70.PubMedGoogle Scholar
- Pugsley AP. The complete general secretory pathway. Microbiol Rev. 1993;57:50–108.PubMedGoogle Scholar
- Fath MJ, Kolter R. ABC transporters: bacterial exporters. Microbiol Rev. 1993;57:997–1017.
- Stephens C, Shapiro L. Targetted protein secretion in bacterial pathogenesis. Curr Biol. 1996;6:927–30. DOIPubMedGoogle Scholar
- Aizawa S-I. Flagellar assembly in Salmonella typhimurium. Mol Microbiol. 1996;19:1–5. DOIPubMedGoogle Scholar
- Harshey RM, Toguchi A. Spinning tails: homologies among bacterial flagellar systems. Trends Microbiol. 1996;4:226–31. DOIPubMedGoogle Scholar
- Kaniga K, Bossio JC, Galán JE. The Salmonella typhimurium invasion genes invF and invG encode homologues of the AraC and PulD family of proteins. Mol Microbiol. 1994;13:555–68. DOIPubMedGoogle Scholar
- Ménard R, Sansonetti PJ, Parsot C. The secretion of the Shigella flexneri Ipa invasins is induced by the epithelial cell and controlled by IpaB and IpaD. EMBO J. 1994;13:5293–302.PubMedGoogle Scholar
- Rosqvist R, Magnusson K, Wolf-Watz H. Target cell contact triggers expression and polarized transfer of Yersinia YopE cytotoxin into mammalian cells. EMBO J. 1994;13:964–72.PubMedGoogle Scholar
- Galán JE. Molecular genetic bases of Salmonella entry into host cells. Mol Microbiol. 1996;20:263–71. DOIPubMedGoogle Scholar
- Straley SC, Perry RD. Environmental modulation of the gene expression and pathogenesis in Yersinia. Trends Microbiol. 1995;3:310–7. DOIPubMedGoogle Scholar
- Lee C, Falkow S. The ability of Salmonella to enter mammalian cells is affected by bacterial growth states. Proc Natl Acad Sci U S A. 1990;89:1847–51. DOIGoogle Scholar
- Hromockyj AE, Tucker SC, Maurelli AT. Temperature regulation of Shigella virulence: identification of the repressor gene virR, an analogue of hns, and partial complementation by tyrosyl transfer RNA. Mol Microbiol. 1992;6:2113–24. DOIPubMedGoogle Scholar
- Pettersson J, Nordfelth R, Dubrinina E, Bergaman T, Gustfsson M, Magnusson KE, Modulation of the virulence factor expression by pathogen target cell contact. Science. 1996;273:1231–3. DOIPubMedGoogle Scholar
- Kenny B, Lai L-C, Finlay BB, Donnenberg MS. EspA, a protein secreted by enteropathogenic Escherichia coli, is required to induce signals in epithelial cells. Mol Microbiol. 1996;20:313–23. DOIPubMedGoogle Scholar
- Salyers AA, Whitt DD. Bacterial Pathogenesis: A Molecular Approac, first ed. Washington D.C.: ASM Press, 1994.
- Cornelis GR. Yersinia Pathogenicity Factors. In: Hormaeche CE, Penn CW, Smyth CJ, editors. Molecular Biology of Bacterial Infection: Current Status and Future Perspectives. Cambridge: Cambridge University Press, 1992.
- Persson C, Nordfelth R, Holmström A, Håkansson S, Rosqvist R, Wolf-Watz H. Cell-surface-bound Yersinia translocate the protein tyrosine phosphatase YopH by a polarized mechanism into the target cell. Mol Microbiol. 1995;18:135–50. DOIPubMedGoogle Scholar
- Håkansson S, Galyov EE, Rosqvist R, Wolf-Watz H. The Yersinia YpkA Ser/Thr kinase is translocated and subsequently targeted to the inner surface of the HeLa cells plasma membrane. Mol Microbiol. 1996;20:593–603. DOIPubMedGoogle Scholar
- Sory M-P, Cornelis GR. Translocation of a hybrid YopE-adenylate cyclase from Yersinia enterocolitica into HeLa cells. Mol Microbiol. 1994;14:583–94. DOIPubMedGoogle Scholar
- Bliska JB, Guan K, Dixon JE, Falkow S. Tyrosine phosphatase hydrolysis of host proteins by an essential Yersinia virulence determinant. Proc Natl Acad Sci U S A. 1991;61:3914–21.
- Ménard R, Dehio C, Sansonetti PJ. Bacterial entry into epithelial cells: the paradigm of Shigella. Trends Microbiol. 1996;4:220–6. DOIPubMedGoogle Scholar
- Jones B, Pascopella L, Falkow S. Entry of microbes into the host: using M cells to break the mucosal barrier. Curr Opin Immunol. 1995;7:474–8. DOIPubMedGoogle Scholar
- Galán JE, Curtiss IR. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc Natl Acad Sci U S A. 1989;86:6386–7. DOIGoogle Scholar
- Shea JE, Hensel M, Gleeson C, Holden DW. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc Natl Acad Sci U S A. 1996;93:2593–7. DOIPubMedGoogle Scholar
- Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269:400–3. DOIPubMedGoogle Scholar
- Rosqvist R, Håkansson S, Forsberg Å, Wolf-Watz H. Functional conservation of the secretion and translocation machinery for virulence proteins of Yersiniae, Salmonellae and Shigellae. EMBO J. 1995;14:4187–95.PubMedGoogle Scholar
- Hermant D, Ménard R, Arricau N, Parsot C, Popoff MY. Functional conservation of the Salmonella and Shigella effectors of entry into epithelial cells. Mol Microbiol. 1995;17:781–9. DOIPubMedGoogle Scholar
- Woestyn S, Allaoui A, Wattiau P. YscN, the putative energizer of the Yersinia Yop secretion machinery. J Bacteriol. 1994;176:1561–9.PubMedGoogle Scholar
- Plano GV, Straley SC. Mutations in yscC, yscD, and yscG prevent high level expression and secretion of V antigen and Yops in Yersinia pestis. J Bacteriol. 1995;177:3843–54.PubMedGoogle Scholar
- Allaoui A, Woestyn S, Sluiters C, Cornelis GR. YscU, a Yersinia enterocolitica inner membrane protein involved in Yop secretion. J Bacteriol. 1994;176:4534–42.PubMedGoogle Scholar
- Fields K, Plano GV, Straley SC. A low-Ca2+ response (LCR) secretion (ysc) locus lies within the lcrB region of the LCR plasmid in Yersinia pestis. J Bacteriol. 1994;176:569–79.PubMedGoogle Scholar
- Allaoui A, Schulte R, Cornelis GR. Mutational analysis of Yersinia enterocolitica virC operon: characterization of yscE,F,G, I, J, K, required for Yop secretion and yscH encoding YopR. Mol Microbiol. 1995;18:343–55. DOIPubMedGoogle Scholar
- Allaoui A, Scheen R, de Rouvroit CL, Cornelis GR. VirG, a Yersinia enterocolitica lipoprotein involved in Ca2+ dependency, is related to ExsB of Pseudomonas aeruginosa. J Bacteriol. 1995;177:4230–7.PubMedGoogle Scholar
- Macnab RM. Flagella and Motility. In: Neidhardt FC et al., editors. Escherichia coli and Salmonella typhimurium Cellular and Molecular Biology. Washington, D. C.: ASM Press, 1996.
- Rimpiläinen M, Forsberg Å, Wolf-Watz H. A novel protein, LcrQ, involved in the low-calcium response of Yersinia pseudotuberculosis shows extensive homology to YopH. J Bacteriol. 1992;174:3355–63.PubMedGoogle Scholar
- Håkansson S, Bergman T, Vanooteghem J-C, Cornelis G, Wolf-Watz H. YopB and YopD constitute a novel class of Yersinia Yop proteins. Infect Immun. 1993;61:71–80.PubMedGoogle Scholar
- Wattiau P, Woestyn S, Cornelis GR. Customized secretion chaperones in pathogenic bacteria. Mol Microbiol. 1996;20:255–62. DOIPubMedGoogle Scholar
- Frithz-Lindsten E, Rosqvist R, Johansson L, Forsberg Å. The chaperone-like protein YerA of Yersinia pseudotuberculosis stabilizes YopE in the cytoplasm but is dispensable for targeting to the secretion loci. Mol Microbiol. 1995;16:635–47. DOIPubMedGoogle Scholar
- Ménard R, Sansonetti P, Parsot C, Vasselon T. Extracellular association and cytoplasmic partitioning of the IpaB and IpaC invasins of S. flexneri. Cell. 1994;79:515–25. DOIPubMedGoogle Scholar
- Skrzypek E, Straley SC. LcrG, a secreted protein involved in negative regulation of the low-calcium response in Yersinia pestis. J Bacteriol. 1993;175:3520–8.PubMedGoogle Scholar
- Forsberg Å, Viitanen A-M, Skurnik M, Wolf-Watz H. The surface-located YopN protein is involved in calcium signal transduction in Yersinia pseudotuberculosis. Mol Microbiol. 1991;5:977–86. DOIPubMedGoogle Scholar
- Hughes KT, Gillen KL, Semon MJ, Karlinsey JE. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science. 1993;262:1277–80. DOIPubMedGoogle Scholar
- Ginocchio C, Galán JE. Functional conservation among members of the Salmonella typhimurium InvA family of proteins. Infect Immun. 1994;63:729–32.
- Jarvis KG, Girón JA, Jerse AE, McDaniel TK, Donnenberg MS, Kaper JB. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc Natl Acad Sci U S A. 1995;92:7996–8000. DOIPubMedGoogle Scholar
- Ochman H, Groisman EA. The evolution of invasion by enteric bacteria. Can J Microbiol. 1995;41:555–61. DOIPubMedGoogle Scholar
- Li J, Ochman H, Groisman EA, Boyd EF, Solomon F, Nelson K, Relationship between evolutionary rate and cellular location among the Inv/Spa invasion proteins of Salmonella enterica. Proc Natl Acad Sci U S A. 1995;92:7252–6. DOIPubMedGoogle Scholar
- Lee CA. Pathogenicity islands and the evolution of bacterial pathogens. Infect Agents Dis. 1996;5:1–7.PubMedGoogle Scholar
- Morschhäuser J, Vetter V, Emödy L, Hacker J. Adhesin regulatory genes within large, unstable DNA regions of pathogenic Escherichia coli: cross-talk between different adhesin gene clusters. Mol Microbiol. 1994;11:555–66. DOIPubMedGoogle Scholar
- Mills DM, Bajaj V, Lee CA. A 40 kb chromosomal fragment encoding Salmonella typhimurium invasion genes is absent from the corresponding region of the Escherichia coli K-12 chromosome. Mol Microbiol. 1995;15:749–59. DOIPubMedGoogle Scholar
- Stein MA, Leung KY, Zwick M, Garcia-del Portillo F, Finlay BB. Identification of a Salmonella virulence gene required for formation of filamentous structures containing lysosomal membrane glycoproteins within epithelial cells. Mol Microbiol. 1996;20:151–64. DOIPubMedGoogle Scholar
- Fetherston JD, Schuetze P, Perry RD. Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 kb of chromosomal DNA which is flanked by a repetitive element. Mol Microbiol. 1992;6:2693–704. DOIPubMedGoogle Scholar
- Fetherston JD, Perry RD. The pigmentation locus of Yersinia pestis KIM6+ is flanked by an insertion sequence and includes the structural genes for pesticin sensitivity and HMWP2. Mol Microbiol. 1994;13:697–708. DOIPubMedGoogle Scholar
- Iteman I, Guiyoule A, de Almeida AMP, Guilvout I, Baranton G, Carniel E. Relationship between loss of pigmentation and deletion of the chromosomal iron-regulated irp2 gene in Yersinia pestis: evidence for separate but related events. Infect Immun. 1993;61:2717–22.PubMedGoogle Scholar
- Rakin A, Urbitsch P, Heeseman J. Evidence for two evolutionary lineages of highly pathogenic Yersinia species. J Bacteriol. 1995;177:2292–8.PubMedGoogle Scholar
- Matic I, Taddei F, Radman M. Genetic barriers among bacteria. Trends Microbiol. 1996;4:69–73. DOIPubMedGoogle Scholar
- Falkow S. The evolution of pathogenicity in Escherichia, Shigella, and Salmonella. In: Neidhardt FC, et al. Escherichia coli and Salmonella Cellular and Molecular Biology. Washington, D. C.: ASM Press, 1996.
- Waldor MK, Mekalanos JJ. Cholera toxin is encoded by a filamentous bacteriophage that uses TCP pili as a receptor. Science. 1996;272:1910–4. DOIPubMedGoogle Scholar
- Guttman DS, Dykhuizen DE. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science. 1994;266:1380–3. DOIPubMedGoogle Scholar
- Inouye S, Sunshine MG, Six EW, Inouye M. Retronphage R73: an E. coli phage that contains a retroelement and integrates into a tRNA gene. Science. 1991;252:969–71. DOIPubMedGoogle Scholar
- Reiter W, Palm P, Yeats S. Transfer RNA genes frequently serve as integration sites for prokaryotic genetic elements. Nucleic Acids Res. 1989;17:1907–14. DOIPubMedGoogle Scholar
- Sun J, Inouye M, Inouye S. Association of a retroelement with a P4-like cryptic prophage (Retronphage R73) integrated into the selenocystyl tRNA gene of Escherichia coli. J Bacteriol. 1991;173:4171–81.PubMedGoogle Scholar
- Akerley BJ, Cotter PA, Miller JF. Ectopic expression of the flagellar regulon alters development of the Bordetella host interaction. Cell. 1995;80:611–20. DOIPubMedGoogle Scholar
- Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschape H, Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect Immun. 1994;62:606–14.PubMedGoogle Scholar
- McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci U S A. 1995;92:1664–8. DOIPubMedGoogle Scholar
- Ritter A, Blum G, Emody L, Kerenyi M, Bock A, Neuhierl B, tRNA genes and pathogenicity islands: influence on virulence and metabolic properties of uropathogenic Escherichia coli. Mol Microbiol. 1995;17:109–21. DOIPubMedGoogle Scholar
- Zagaglia C, Casalino M, Colonna B, Conti C, Calconi A, Nicoletti M. Virulence plasmids of enteroinvasive Escherichia coli and Shigella flexneri integrate into a specific site on the host chromosome: integration greatly reduces expression of plasmid-carried virulence genes. Infect Immun. 1991;59:792–9.PubMedGoogle Scholar
- Colonna B, Casalino M, Fradiani PA, Zagaglia C, Naitza S, Leoni L, H-NS regulation of virulence gene expression in enteroinvasive Escherichia coli harboring the virulence plasmid integrated into the host chromosome. J Bacteriol. 1995;177:4703–12.PubMedGoogle Scholar
- Zsigray RM, Hopper JB, Zukowski K, Chesbro WR. Integration of the Vwa plasmid into the chromosome of Yersinia pestis strains harboring F' plasmids of Escherichia coli. Infect Immun. 1985;47:670–3.PubMedGoogle Scholar
- Zsigray RM, Lawton WD, Surgalla MJ. Repression of the virulence of Yersinia pestis by an F' plasmid. Infect Immun. 1983;39:974–6.PubMedGoogle Scholar
- Protsenko OA, Filippov AA, Kutyrev VV. Integration of the plasmid encoding the synthesis of capsular antigen and murine toxin into Yersinia pestis chromosome. Microb Pathog. 1991;11:123–8. DOIPubMedGoogle Scholar
- Whittam TS, Wolfe ML, Wachsmuth IK, Ørskov F, Ørskov I, Wilson RA. Clonal relationships among Escherichia coli strains that cause hemorrhagic colitis and infantile diarrhea. Infect Immun. 1993;61:1619–29.PubMedGoogle Scholar
- Waldor MK, Mekalanos JJ. Vibrio cholerae O139 specific gene sequences. Lancet. 1994;343:1366. DOIPubMedGoogle Scholar
- Pajni S, Charu S, Bhasin N, Ghosh A, Ramamurthy T, Nair GB, Studies on the genesis of Vibrio cholerae O139: identification of probable progenitor strains. Journal of Molecular Microbiology. 1995;42:20–5. DOIGoogle Scholar
- Bik EM, Bunschoten AE, Gouw RD, Mooi FR. Genesis of the novel epidemic Vibrio cholerae 139 strain: evidence for horizontal transfer of genes involved in polysaccharide synthesis. EMBO J. 1995;14:209–16.PubMedGoogle Scholar
- Sory M-P, Hermand P, Vaerman J-P, Cornelius GR. Oral immunization of mice with a live recombinant Yersinia enterocolitica O:9 strain that produces the cholera toxin B subunit. Infect Immun. 1990;58:3830–6.
- Falkow S, Small P, Isberg R, Hayes SF, Corwin D. A molecular strategy for the study of bacterial invasion. Rev Infect Dis. 1987;9:S450–5.PubMedGoogle Scholar
- Swenson DL, Bukanov NO, Berg DE, Welch RA. Two pathogenicity islands in uropathogenic Escherichia coli strain J96: cosmid cloning and sample sequencing. Infect Immun. 1996;64:3736–43.PubMedGoogle Scholar
- Blum G, Falbo V, Caprioli A, Hacker J. Gene clusters encoding the cytotoxic necrotizing factor type 1, prs-fimbriae and -hemolysin form the pathogenicity island II of the uropathogenic Escherichia coli strain J96. Federation of European Microbiological Societies Microbiology Letters. 1995;126:189–96.
- Comstock LE, Johnson JA, Michalski JM, Morris JG Jr, Kaper JB. Cloning and sequence of a region encoding a surface polysaccharide of Vibrio cholerae O139 and characterization of the insertion site in the chromosome of Vibrio cholerae 01. Mol Microbiol. 1996;19:815–26. DOIPubMedGoogle Scholar
- Gouin E, Mengaud J, Cossart P. The virulence gene cluster of Listeria monocytogenes is also present in Listeria ivanovii, an animal pathogen, and Listeria seeligeri, a nonpathogenic species. Infect Immun. 1994;62:3550–3.PubMedGoogle Scholar
Figure
Tables
Cite This ArticleTable of Contents – Volume 2, Number 4—October 1996
| EID Search Options |
|---|
|
|
|
|
|
|