Volume 13, Number 10—October 2007
Research
Evolutionary Relationships between Bat Coronaviruses and Their Hosts
Abstract
Recent studies have suggested that bats are the natural reservoir of a range of coronaviruses (CoVs), and that rhinolophid bats harbor viruses closely related to the severe acute respiratory syndrome (SARS) CoV, which caused an outbreak of respiratory illness in humans during 2002–2003. We examined the evolutionary relationships between bat CoVs and their hosts by using sequence data of the virus RNA-dependent RNA polymerase gene and the bat cytochrome b gene. Phylogenetic analyses showed multiple incongruent associations between the phylogenies of rhinolophid bats and their CoVs, which suggested that host shifts have occurred in the recent evolutionary history of this group. These shifts may be due to either virus biologic traits or host behavioral traits. This finding has implications for the emergence of SARS and for the potential future emergence of SARS-CoVs or related viruses.
Severe acute respiratory syndrome (SARS) emerged in November 2002 in southern People’s Republic of China (1), and a SARS coronavirus (SARS-CoV) was identified as the etiologic agent (2). These events and the identification of SARS-CoV in animals associated with the wildlife trade in southern China (3) have led to a rapid resurgence of interest in CoVs of different origins. This resurgence led to discovery of 2 novel human CoVs (4,5); identification of SARS-like CoVs in horseshoe bats (Rhinolophus macrotis, R. ferrumequinum, R. pearsoni, and R. sinicus) (6,7); and identification of other CoVs in bat species (R. sinicus, R. ferrumequinum, Miniopterus magnater [M. magnater has been misidentified as M. schreibersi (8) in reports on SARS-like CoV], Pipistrellus abramus, P. pipistrellus, Tylonycteris pachypus, Myotis ricketti, and Scotophilus kuhlii) (7,9–12). However, evolutionary relationships among these CoVs and their bat hosts have not been examined.
Studies in species other than bats have examined host-virus phylogeny and identified coevolutionary relationships (13–16) or incongruous phylogenetic patterns (17). These findings suggest recent pathogen host shifts (defined as interspecies transmission followed by establishment and long-term persistence in the new host species [18]). Other studies have demonstrated that the relationship between viral phylogeny and geographic location and identification hosts (viral phylogeography [19]) can yield information on the origin of emerging zoonoses (19,20).
Knowing the high genetic diversity of bat CoVs, we carried out a systematic phylogenetic study of the viruses and their hosts to examine evolutionary relationships between bat CoVs and bats. The aim was to further investigate the origin of SARS-like CoVs and SARS. Our results suggest host-pathogen divergence and host shifts in the recent evolutionary history of these viruses and their hosts. We discuss host behavioral traits and viral traits that might have given rise to these patterns and comment on the implications of our findings for the emergence of SARS-CoV.
CoV Sequences
Only CoVs from bats were included in this study. We used gene sequences that Tang et al. obtained from 10 bat species (R. sinicus, R. ferrumequinum, R. macrotis, R. pearsoni, M. magnater, P. abramus, P. pipistrellus, T. pachypus, S. kuhlii, and Myotis ricketti) (10). An additional 57 bat CoV sequences available in GenBank were also included in this analysis.
Bat Mitochondrial Cytochrome b (cyt b) Gene Sequences
Tissue samples were obtained from 3-mm wing membrane biopsy specimens from wild bats, which had been caught in 9 provinces of China, that had been preserved in 99% ethanol. Genomic DNA was extracted by using the DNeasy Tissue Kit (QIAGEN, Valencia, CA, USA) and stored at –20°C. We used complete cyt b sequences of R. ferrumequinum, P. abramus, and P. pipistrellus, which have recently been published and are available in Genbank. We generated cyt b sequences from M. magnater (n = 4), T. pachypus (n = 3), R. macrotis (n = 2), R. pearsoni (n = 2), R. sinicus (n = 2), S. kuhlii (n = 1), and Myotis ricketti (n = 1).
PCR mixtures were prepared in 50-μL volumes containing 25 μL 2× EXTaq DNA polymerase (TaKaRa, Kyoto, Japan). Two pairs of primers, Bat_Cytb_1 (5′-TAG AAT ATC AGC TTT GGG TG-3′) (21) with Bat_Cytb_2 (5′-AAA TCA CCG TTG TAC TTC AAC-3′) (21), and Bat_Cytb_2 with BAT15R (5′-TCA GCT TTG GGT GTT GAT GG-3′) (22), were used because of amplification specificity of certain primers in some species. Amplification was conducted at an initial denaturing temperature at 94°C for 30 s; 34 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 90s; and a final extension at 72°C for 10 min. The PCR samples were then stored at 4°C. The complete mitochondrial cyt b gene (1,140 bp) was amplified and sequenced. These sequences have been submitted to GenBank and accession numbers are shown in the Table.
Phylogenetic Analysis of CoV Sequences
Figure 1
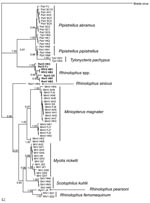
Figure 1. Phylogram of bat coronaviruses based on the 440-bp RNA-dependent RNA polymerase gene region. Methods used are described in the text. Values to the left of branches are Bayesian posterior probabilities. Scale...
For virus phylogeny studies, sequences from a 440-bp fragment of the RNA-dependent RNA polymerase (RdRp) gene, which is highly conserved among different CoVs, were obtained and analyzed (10). Multiple alignments of the 440-bp RdRp partial sequence of bat CoVs were conducted in ClustalX version 1.81 (23). Bayesian analyses were conducted with MrBayes version 3.1.2 (24). Neighbor-joining analyses (with the Jukes-Cantor model) were used to validate the Bayesian result in MEGA3 (25). A total of 67 unique CoV sequences (Figure 1) were analyzed with MrBayes version 3.1.2 in the generalized time reversible model of evolution as determined by the Akaike Information Criterion in MODELTEST version 3.7 (26). Four consecutive Metropolis-coupled Markov chain Monte Carlo computations were run for 2 million generations, with trees sampled every 100 generations. Initial trees were random. On the basis of stabilization of preliminary runs, the first 3,000 trees were discarded before generation of the consensus tree. The Bayesian consensus tree was rooted to Breda virus (AY427798), a related CoV (Figure 1).
Phylogenetic Analyses of Bat cyt b Gene Sequences
For bat phylogeny, we used the complete mitochondrial cyt b gene to construct maximum likelihood (ML) and Bayesian phylograms. The cyt b sequence data were aligned by using ClustalX version 1.81 as above. ML analysis was performed by using PAUP* version 4.0b (27). The most appropriate substitution model (generalized time reversible + Γ + I) with the parameters matrix = 0.4835 × 9.6665 × 0.3815 × 0.2973 × 7.1418, base frequency = 0.3576 × 0.3420 × 0.0748, rates = gamma, shape = 0.6008, and proportion of invariable sites unable to accept substitutions = 0.4078 for ML and subsequent Bayesian analysis was calculated by using the program Modeltest 3.7 (26). We used heuristic searches (10 replicates, random addition of taxa, with tree bisection and reconnection branch swapping), followed by 100 bootstrap iterations for robustness of the ML tree. Bayesian analysis was also used to construct a tree with 4 simultaneous Markov chains for 1 million generations. Trees were sampled every 20 generations, and the first 5,000 trees were discarded before the consensus tree was made (on the basis of practical values of stabilizing likelihood).
Genetic Diversity among Bats and CoVs
We compared the genetic diversity of CoVs isolated from rhinolophids and vespertilionids and the corresponding diversity among bat taxa by using the index of nucleotide diversity (π) described by Nei (28) in Arlequin version 3.1 (29). Analyses were performed on uncorrected pairwise genetic distances between sequences.
Figure 2
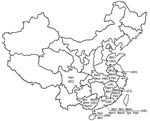
Figure 2. Distribution of coronaviruses isolated in the People’s Republic of China. RsV, detected in Rhinolophus sinicus; PaV, detected in Pipistrellus abramus; TpV, detected in Tylonycteris pachypus; RfV, detected in R. ferrumequinum; RmV,...
By combining information derived from the phylogram of bat CoVs, together with data on the geographic origin of viruses, we were able to describe the phylogeographic distributions for known CoVs from bats in China (Figures 1, 2; Table). Bat SARS-like CoVs formed a monophyletic clade. Species-specific host restriction was found for CoVs in 4 of 7 bats species (Myotis ricketti, M. magnater, P. abramus, and T. pachypus) sampled from >1 geographic location, and these clustered with high Bayesian posterior probability. Overall phylogenetic relationships between virus lineages were similar across our analyses, and well-supported genetic structure was observed within some CoV lineages. For example, CoVs isolated from M. magnater were monophyletic but formed 3 well-supported clades with no evidence of geographic structure (Bayesian posterior probability [PP] = 1.0 for each). A similar pattern was apparent in CoVs from Myotis ricketti, which formed 2 geographically overlapping independent clades (PP = 0.99 and 1.0, respectively). One T. pachypus was infected by a virus that clustered with moderate statistical support (PP = 0.91) within the larger clade associated with P. abramus, which indicated a potential interspecies transmission event or recent evolutionary host shift (defined as interspecies transmission followed by establishment and long-term persistence in the new host species [18]) (Figure 1).
Figure 3

Figure 3. Phylogenetic relationships between coronaviruses (left) and their host bat species added for reference (right). Abbreviations on both sides denote viruses harbored by bats (marked as V on the left) and bats...
Figure 4
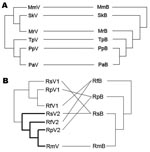
Figure 4. Phylogenetic relationships between coronaviruses (CoVs) (left) and bats (right) in the A) Vespertilionidae and B) Rhinolophidae. Abbreviations on both sides denote viruses harbored by bats (marked as V on the left)...
Phylograms of host sequences were also constructed and were essentially of the same topology with high support whether derived by using MrBayes version 3.1.2 or MEGA3 (data not shown). When we mapped host phylogram to virus, virus phylogeny did not always track host phylogeny (Figure 3). When separate host-virus phylograms were constructed for the 2 bat families (Verspertilionidae and Rhinolophidae), different corresponding relationships were evident. Verspertilionids and their CoVs showed phylogenetic congruence, and rhinolophids and their CoVs showed incongruous phylogenies (Figure 4).
We found evidence for evolutionarily divergent relationships for some vespertilionid viruses and their hosts when analyzed at the family scale (Figure 4, panel A). For example, divergence between viruses harbored by P. pipistrellus and P. abramus is congruent with their hosts. The divergence among other viruses was incongruent with divergence of host species, e.g., those from S. kuhlii and Myotis ricketti.
Rhinolophid bats and their viruses were analyzed at a different taxonomic scale (within genus). In this co-phylogeny, viral host shifts were the evident virus-host feature (Figure 4, panel B). Except for R. macrotis, all rhinolophidae bats had 2 distinct lineages of CoVs, and host shifts were found among viruses carried by R. ferrumequinum, R. pearsoni, and R. sinicus.
Genetic diversity of CoVs harbored by rhinolophids and vespertilionids was similar (vespertilionids π = 0.27 ± 0.13; rhinolophids π = 0.25 ± 0.13). In contrast, genetic diversity of cyt b sequences from bats was much higher among the vespertilonids (π = 0.17 ± 0.007) than among the rhinolophids (π = 0.09 ± 0.006).
CoVs sequenced from different bats of the same species clustered together, even when bats were collected in locations 1,000–2,000 km apart. This pattern was found for CoVs from P. abramus, T. pachypus, Myotis ricketti, and M. magnater. Bats of the genus Miniopterus are known to migrate long distances (30), which explains why the phylogeny of viruses isolated from M. magnater sampled in distant places (Guangxi, Anhui, Fujian, and Hong Kong) lacks geographic structure. In nonmigrating species such as bats of the genera Pipistrellus and Tylonycteris, intimate physical contact of bats in same cave or the same bamboo roost site, as well as periodic exchange of bats among neighboring colonies, may facilitate virus transmission among populations.
Despite the co-roosting of many bats species, we found little evidence of host shifts for some viruses. For example, CoVs from M. magnater and Myotis ricketti sampled in the same cave in Guangxi were divergent, although sample size was limited. Although Myotis ricketti has a closer phylogenetic relationship with T. pachypus, P. pipistrellus, and P. abramus than with M. magnater and S. kuhlii, its behavior and habits are closer to those of the last group. For example, Myotis ricketti and S. kuhlii bats roosts in caves (although S. kuhlii also roosts under palm leaves), whereas T. pachypus roosts inside bamboo and P. abramus roosts almost entirely in old buildings. Thus, it seems plausible that the close phylogenetic relationship between viruses harbored by Myotis ricketti and S. kuhlii reflects the similar behavior and ecology of their hosts.
The phylogenetic and phylogeographic associations we found suggest that there may be a coevolutionary relationship between some bat CoVs and their hosts. For example, sister taxa within the genus Pipistrellus independently maintained 2 distinct viruses that share a most recent common ancestor. A similar relationship was apparent among the viruses of some closely related genera (e.g., Pipistrellus and Tylonycteris), whereby divergence of each genus was mirrored by divergence in viral phylogeny. However, viruses are usually thought to have evolved more recently than their hosts (31). Thus, apparent coevolutionary patterns may reflect either a high frequency of host shift among closely related bat species or simultaneous lineage splitting of hosts and viruses. Host shifts among related bats might be favored by a variety of mechanisms, including preadaptation to overcome immune defenses or greater rates of interspecific contact relative to distantly related bat species. Phylograms with better resolution would enable statistical comparison of phylogenetic congruence and estimation of divergence times.
In the vespertilionids, close phylogenetic concordance between host and virus suggests a close, possibly evolutionarily divergent relationship. However, there are different scales of comparison between the Vespertillionidae, in which all but 1 CoV came from separate genera, and the Rhinolophidae, in which we examined a co-phylogeny of multiple species within 1 genus. Genetic diversity in the vespertilionids sampled was nearly double that of the rhinolophids, which was probably due to the greater number of species sampled and their broader taxonomic range. Despite this greater genetic diversity among vespertilionid bat hosts, the genetic diversity of CoVs did not differ between vespertilionids and rhinopholids. This diversity suggests that vespertilionids may maintain undiscovered CoVs or that rhinolophids might harbor disproportionate CoV diversity relative to diversity of their genus. We propose that future work may identify more vespertilionid bat CoVs, which would enable an accurate comparison of propensity for host shifts within this group.
In the rhinolophids, the host phylogram demonstrated genetic divergence between R. ferrumequinum and other species, as shown by the division of Rhinolophus bats into 2 groups. Each of these groups harbors CoVs from 2 clusters (SARS-like CoVs and other CoVs), which suggests multiple introductions of CoVs into these species.
Lack of concordance between phylogenies of rhinolophid bats and their CoVs can be interpreted as evidence for host shifts between bats of the genus Rhinolophus. Different species of Rhinolophus are often observed roosting inside the same cave, which facilitates virus transmission between species. However, the degree of host shifting of rhinolophid bat CoVs may not be particularly high relative to other genera of bats. This observation will be clarified when a greater diversity of CoVs from other bat genera is reported and the sequences are analyzed. These requirements support the need for further research on bat viruses (32,33).
Host-shifting within the genus Rhinolophus would likely be promoted if these bats shed CoVs in a way that makes them more available to other Rhinolophus spp.; had behavioral traits that lead to increased contact with other Rhinolophus spp.; or if CoVs harbored by these bats have structural, biologic, or other traits that make them more readily transmitted to other Rhinolophus spp. Two lines of evidence suggest that host traits are the most parsimonious explanation for host shifts within the genus Rhinolophus. First, SARS-like CoVs and other rhinolophid CoVs (RfV1 and RpV1) show evidence of interspecies transmission. Second, CoVs from other bat groups that are phylogenetically much closer to RfV1 and RpV1 than to the SARS-like CoVs do not show evidence of successful host shifts. Thus, the ability to jump hosts is unlikely to be a strictly viral trait.
The phylogeography of bat CoVs suggests that the bat SARS-like CoVs form a monophyletic clade that is both phylogenetically distinct from other bat CoVs and geographically isolated. Although we acknowledge that this interpretation may be limited by sample size, it may also indicate that rhinolophid bats, the hosts of a cluster of SARS-like CoVs within which human and civet SARS CoV nestle phylogenetically (6,7), are more likely to foster the host shifts of CoVs than are other bat species. The potential for close contact between bats, civets, and humans in the wildlife trade in southern China, coupled with a possible propensity of these bats to foster CoV host-shifts, could explain SARS-like CoVs as the source of SARS-CoV. This potential supports molecular results on bat CoVs that suggest a recent host shift from bats to civets or other animals and humans (34). Such host shifts may indicate a risk posed by other bat CoVs for novel disease emergence. Finally, the ability of SARS-like CoVs to be transmitted between and establish in new species (i.e., to undergo host shifts) is consistent with other CoVs. This has been shown for several CoVs of livestock species (35) and has been used to support their inclusion as 1 of the groups of viruses most likely to be responsible for emerging zoonoses, even before the emergence of SARS (36).
The total diversity of CoVs (including SARS-like CoVs) in bats has likely not been fully described. The genus Rhinolophus (8) contains 77 bat species distributed in Asia, Europe, and Africa. The recent discoveries of bat CoVs in the United States (37) and SARS-COVs in African bats (38) support the hypothesis that CoVs are diverse and widespread in bat species. Given the diversity of CoVs in this group, and their propensity for host shifts, further viral discovery in rhinolophids may assist in understanding and ultimately controlling the emergence of zoonotic viruses. Bats are increasingly recognized as reservoirs of many highly lethal zoonotic agents (32). Understanding their diversity, behavior, and mechanisms of virus transmission may play a key role in preventing future outbreaks of both known and unknown zoonotic diseases of bat origin.
Mr Cui is a doctoral candidate at the School of Life Science, East China Normal University, Shanghai. His research interests include coevolution of viruses and hosts and bat-related viral epidemiology.
Acknowledgment
This study was supported by the State Key Program for Basic Research grant (2005CB523004) to Z.S. and S.Z.; project Animal Reservoir of SARS-CoV from the Ministry of Science and Technology of China to Y.G. and S.Z.; the Sixth Framework Program EPISARS from the European Commission (no. 51163) to Z.H., G.Z., and S.Z.; the Australian Biosecurity Cooperative Research Centre (2.026RE) to L.W. and H.E.F.; a National Institutes of Health/National Science Foundation Ecology of Infectious Diseases award from the John E. Fogarty International Center (R01-TW05869) to H.E.F. and P.D.; and an award to the Consortium for Conservation Medicine from the V. Kann Rasmussen Foundation.
References
- Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Bercker S, Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–76. DOIPubMedGoogle Scholar
- Kuiken T, Fouchier RA, Schutten M, Rimmelzwaan GF, van Amerongen G, van Riel D, Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362:263–70. DOIPubMedGoogle Scholar
- Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–8. DOIPubMedGoogle Scholar
- van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolther KC, Identification of a new human coronavirus. Nat Med. 2004;10:368–73. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79:884–95. DOIPubMedGoogle Scholar
- Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–9. DOIPubMedGoogle Scholar
- Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040–5. DOIPubMedGoogle Scholar
- Simmons NB. Order Chiroptera. In: Wilson DE, Reeder DM, editors. Mammal species of the world. Baltimore: Johns Hopkins University Press; 2005. p. 312–529.
- Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, Identification of a novel coronavirus in bats. J Virol. 2005;79:2001–9. DOIPubMedGoogle Scholar
- Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, Prevalence and genetic diversity of coronaviruses in bats from China. J Virol. 2006;80:7481–90. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Li KS, Poon RW, Wong BH, Tsoi HW, Molecular diversity of coronaviruses in bats. Virology. 2006;351:180–7. DOIPubMedGoogle Scholar
- Chu DK, Poon LL, Chan KH, Chen H, Guan Y, Yuen KY, Coronaviruses in bent-winged bats (Miniopterus spp.). J Gen Virol. 2006;87:2461–6. DOIPubMedGoogle Scholar
- Lukashov VV, Goudsmit J. Evolutionary relationships among parvoviruses: virus-host coevolution among autonomous primate parvoviruses and links between adeno-associated and avian parvoviruses. J Virol. 2001;75:2729–40. DOIPubMedGoogle Scholar
- Kariwa H. Bunyavirus virus and host relationship: the coevolution between hantavirus and rodent. Uirsu. 2002;52:61–7.
- Herniou EA, Olszewski JA, O’Reilly DR, Cory JS. Ancient coevolution of baculoviruses and their insect hosts. J Virol. 2004;78:3244–51. DOIPubMedGoogle Scholar
- Perez-Losada M, Christensen RG, McClellan DA, Adams BJ, Viscidi RP, Demma JC, Comparing phylogenetic codivergence between polyomaviruses and their hosts. J Virol. 2006;80:5663–9. DOIPubMedGoogle Scholar
- Page RD. Parallel phylogenies: reconstructing the history of host-parasite assemblages. Cladistics. 1994;10:155–73. DOIGoogle Scholar
- Antonovics J, Hood M, Partain J. The ecology and genetics of a host shift: Microbotryum as a model system. Am Nat. 2002;160:S40–53. DOIPubMedGoogle Scholar
- Chen H, Smith G, Li KS, Wang J, Fan XH, Rayner JM, Establishment of multiple sublineages of H5N1 influenza virus in Asia: implications for pandemic control. Proc Natl Acad Sci U S A. 2006;103:2845–50. DOIPubMedGoogle Scholar
- Li G, Jones G, Rossiter SJ, Chen S, Parson S, Zhang S. Phylogenetics of small horseshoe bats from East Asia based on mitochondrial DNA sequence variation. J Mammal. 2006;87:1234–40. DOIGoogle Scholar
- Irwin DM, Kocher TD, Wilson AC. Evolution of the cytochrome b gene of mammals. J Mol Evol. 1991;32:128–44. DOIPubMedGoogle Scholar
- Thompson JD, Gibson TJ, Plewniak F, Jeanmouqin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82. DOIPubMedGoogle Scholar
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5. DOIPubMedGoogle Scholar
- Kumar S, Tamura K, Nei M. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–63. DOIPubMedGoogle Scholar
- Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8. DOIPubMedGoogle Scholar
- Swofford DL. PAUP* beta version: phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland (MA): Sinauer Associates; 2002.
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987.
- Excoffier L, Laval G, Schneider S. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50.PubMedGoogle Scholar
- Miller-Butterworth CM, Jacobs DS, Harley EH. Strong population substructure is correlated with morphology and ecology in a migratory bat. Nature. 2003;424:187–91. DOIPubMedGoogle Scholar
- Holmes EC. Error thresholds and the constraints to RNA virus evolution. Trends Microbiol. 2003;11:543–6. DOIPubMedGoogle Scholar
- Dobson AP. What links bats to emerging infectious diseases? Science. 2005;310:628–9. DOIPubMedGoogle Scholar
- Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–45. DOIPubMedGoogle Scholar
- Vijaykrishna D, Smith GJ, Zhang JX, Peiris JS, Chen H, Guan Y. Evolutionary insights into the ecology of coronaviruses. J Virol. 2007;81:4012–20. DOIPubMedGoogle Scholar
- Saif LJ. Animal coronaviruses: what can they teach us about the severe acute respiratory syndrome? Rev Sci Tech. 2004;23:643–60.PubMedGoogle Scholar
- Burke DS. The evolvability of emerging viruses. In: Nelson AM, Horsburgh CR, editors. Pathology of emerging infections. Washington: American Society for Microbiology; 1998. p. 1–12.
- Dominguez SR, O’Shea TJ, Oko LM, Holmes KV. Detection of group 1 coronaviruses in bats in North America. Emerg Infect Dis. 2007;13:1295–300.PubMedGoogle Scholar
- Müller MA, Paweska JT, Leman PA, Drosten C, Grywna K, Kemp A, Coronavirus antibodies in African bat species. Emerg Infect Dis. 2007;13:1367–70.PubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 13, Number 10—October 2007
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Addresses for correspondence: Shuyi Zhang, School of Life Science, East China Normal University, Shanghai 200062, People’s Republic of China; ; and Peter Daszak, Consortium for Conservation Medicine, 460 W 34th St, New York, NY 10001, USA;
Top