Volume 19, Number 10—October 2013
Research
Immunogenic Mycobacterium africanum Strains Associated with Ongoing Transmission in The Gambia
Cite This Article
Citation for Media
Abstract
In West Africa, Mycobacterium tuberculosis strains co-circulate with M. africanum, and both pathogens cause pulmonary tuberculosis in humans. Given recent findings that M. tuberculosis T-cell epitopes are hyperconserved, we hypothesized that more immunogenic strains have increased capacity to spread within the human host population. We investigated the relationship between the composition of the mycobacterial population in The Gambia, as measured by spoligotype analysis, and the immunogenicity of these strains as measured by purified protein derivative–induced interferon-γ release in ELISPOT assays of peripheral blood mononuclear cells. We found a positive correlation between strains with superior spreading capacity and their relative immunogenicity. Although our observation is true for M. tuberculosis and M. africanum strains, the association was especially pronounced in 1 M. africanum sublineage, characterized by spoligotype shared international type 181, which is responsible for 20% of all tuberculosis cases in the region and therefore poses a major public health threat in The Gambia.
Tuberculosis (TB), caused by bacterial pathogens of the Mycobacterium tuberculosis complex (MTBC), is a major global health problem. Sub-Saharan Africa has the highest rate of TB per capita and the lowest case detection rate; although TB incidence is decreasing globally, incidence rates are increasing in most countries in the West Africa region (1). Moreover, almost half of all TB cases in West Africa are caused by infection with an unusual member of the MTBC, M. africanum, a lineage found exclusively in this region. Although M. africanum was initially described in Senegal in 1968 (2), and despite its importance and high prevalence in this region, relatively little is known about the bacterium (3). In general, M. africanum can be divided into 2 lineages: Afri_1, by SpolDB4 definition (4), corresponding to the green lineage 6 (5), which has the highest prevalence in Senegal, Mali, The Gambia, Guinea-Bissau, and Sierra Leone (3); and Afri_2 (4), corresponding to the brown lineage 5 (5), which is mainly found in the eastern part of West Africa, in countries such as Côte d’Ivoire, Ghana, Benin, Nigeria, and Cameroon (3).
Although transmission of M. africanum from host to host is a crucial element of the spread of the disease, the underlying biological mechanisms triggering transmission are elusive. We assessed transmission dynamics and interaction between the 2 mycobacterial populations in The Gambia, a country in western West Africa, and compared the local situation with previously published data from Guinea-Bissau, another country within the region (6). In particular, considering a recent publication suggesting that conserved mycobacterial T-cell epitopes may play a role in the transmission of the mycobacteria within the host population (7), we investigated whether differences in immunogenicity between M. tuberculosis and M. africanum strains (especially of the predominant Euro-American [EA] and Afri_1 lineages) could predict the success of certain sublineages to transmit and establish themselves within the human host population.
Study Population and Sample Collection
Data for our study came from an ongoing TB case–contact study at the Medical Research Council in The Gambia; TB case-patients were recruited for that study during June 20, 2002–December 21, 2009. Consecutive patients were included after written informed consent if they were >15 years old, resided in the study area (Greater Banjul area), and produced 2 sputum samples that were positive for acid-fast bacilli by Ziehl-Neelsen staining.
Spoligotyping and Analysis
Genomic DNA was purified from the collected sputum samples by using the cetyl trimethyl ammonium bromide and chloroform method, as described (8). Spoligotyping was performed by using commercially available membranes (Ocimum Biolsolutions, Hyderabad, India), according to standardized protocols (9). Spoligotype patterns with ambiguous signature were confirmed by using long-sequence polymorphism PCR.
In addition to analyzing the samples collected in The Gambia, we reanalyzed spoligotypes from published studies from Guinea-Bissau (6). The shared international type (SIT) number was assigned by using the SITVIT database on the Institute Pasteur de Guadeloupe website (www.pasteur-guadeloupe.fr/tb/bd_myco.html). Lineages of spoligotypes were assigned according to SpolDB4 classification by using the TB Lineage online platform (http://tbinsight.cs.rpi.edu/about_tb_lineage.html) (10). Spoligotype data were further analyzed by using spolTools (www.emi.unsw.edu.au/spolTools) (11), which provides online tools for the construction of Spoligoforests (12) and to Detect Emerging Strains of Tuberculosis by Using Spoligotyping (DESTUS) (13) from spoligotype data. As recommended by the provider, the Spoligoforests and DESTUS programs were run with the default settings.
To analyze temporal clustering of spoligotypes from The Gambia, we used SaTScan version 9.1.1 software (www.satscan.org) (14); we conducted a purely temporal analysis for high rates of clustering in a discrete Poisson model. Hospital admission dates for each patient were used as input dates for each spoligotype, and the resolution of the analysis was set to days.
PPD-ELISPOT
Purified protein derivative (PPD) ELISPOT assays were performed as described (15) on a subset of 372 study samples. Quantitative results were expressed as the number of spot-forming units (sfu) that produce interferon-γ in response to M. tuberculosis PPD antigen. Positive wells were predefined as containing >10 sfu more than, and at least 2 times as many as, negative control wells. The negative control well was required to have <30 sfu.
Statistical Analysis
Odds ratios (ORs) and 95% CIs were calculated for analysis of cross-tabulations. To estimate differences between groups, the χ2 test was applied with 2-tailed p values. To confirm the results and to use a more accurate test for 2 × 2 tables, we also performed Fisher exact testing. We considered test results with p values <0.05 to be statistically significant.
The recent transmission index (RTIn–1) was calculated as described (16). Patients with singleton strains were considered to have TB from reactivation and not recent transmission and, therefore, RTIn–1 = 0. The average PPD response of patients with singleton strains was considered the baseline. Following calculation of the PPD response of the singleton strains, all recently transmitted strains with a cluster size of 2 were included, and the average PPD response and RTIn–1 was re-calculated. This procedure was continued by stepwise inclusion of the next bigger genotypic cluster (i.e., [singleton] + [cluster n = 2] + [cluster n = 3]; [singleton] + [cluster n = 2] + [cluster n = 3] + [cluster n = 4]; ...), and recalculation of the average PPD response for each respective RTIn–1 group was performed.
Population Structure of MTBC and Transmission of Isolates
Figure 1
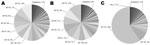
Figure 1. . Spoligotyping results showing population structure of Mycobacterium tuberculosis and M. africanum, The Gambia, 2002–2009. A) All M. tuberculosis sensu stricto lineages (including Euro-American); B) Euro-American lineage; C) M. africanum lineages...
For the study period, 1,003 smear-positive TB cases were identified. Spoligotypes could be obtained from 884 (88%) isolates; many of the strains collected belonged to the M. africanum Afri_1 or M. tuberculosis sensu stricto lineages, and 86% of all M. tuberculosis strains were part of the EA lineage. Therefore, for the remainder of the analysis, we compared all M. tuberculosis sensu stricto isolates (including EA), EA lineage isolates separately, and M. africanum Afri_1 isolates. For the 2 major lineages (Afri_1 and EA), we identified 17–19 genotypes per 100 isolates, of which 9%–12% were found only once as singletons and thus were most likely the result of reactivation of previous disease (Table 1; Figure 1). The remaining spoligotypes (88%–91%) could be assigned to genotypic clusters with an average size of 11.8 and 13.3 isolates for M. tuberculosis EA and M. africanum Afri_1, respectively (Table 1). Assuming that recent transmission was correlated with cluster size and that each cluster contained 1 index case, the RTIn–1 for both populations indicated that 80%–83% of TB cases were attributable to newly acquired infections.
Despite the similarities between the 2 mycobacterial populations, their individual compositions differed drastically. Whereas 59% of the M. africanum population was represented by only a single spoligotype, SIT 181, comprising 198 isolates, the same proportion of the M. tuberculosis EA population contained as many as 11–12 smaller clusters of comparable sizes; with SIT 53 being the largest cluster (Figure 1). Consequently, the θ variable, a maximum-likelihood estimate of the genetic diversity of a population (11), was lower for M. africanum (θ = 22.7) than for M. tuberculosis EA (θ = 27.0) or for all M. tuberculosis sensu stricto (θ = 43.3). The effect of SIT 181 on the population structure became especially apparent when this cluster was excluded from the analysis, resulting in multiple changes to M. africanum population parameters, such as clustering rate, cluster size, and genetic diversity θ (Table 1). The drop in RTI also demonstrates the contribution of SIT 181 to recent transmission within the M. africanum Afri_1 lineage.
Spoligoforests of MTBC in West Africa
Using spoligoforests to display mycobacterial populations (12) takes the genetic relatedness of spoligotypes into consideration and allows deduction of relationships among bacterial sublineages. When we analyzed the 884 MTBC isolates from The Gambia, we found SIT 53 not only to be the most ancestral M. tuberculosis spoligotype but also to constitute the largest M. tuberculosis cluster (Technical Appendix Figure, panel A). Besides this ancestral strain, we identified 4 more recent major spoligotype clusters (SITs 42, 47, 50, and 61) and 5 third-generation clusters (SITs 20, 60, 62, 144, and 183). Most strains belonged to the modern M. tuberculosis lineages. Moreover, when the distribution and size of these individual clusters was considered, M. tuberculosis strains seemed to spread evenly within the host population, resulting in a uniformly distributed structure of the bacterial population. In contrast, the M. africanum population was highly skewed toward a central cluster of SIT 181, next to which (with the exception of SIT 187) no notable secondary spoligotype clusters emerged. Therefore, the population was concentrated around this spoligotype, and most cases of recently transmitted disease could be attributed to this genotype. Similar results were found when we reanalyzed a published spoligotype dataset from Guinea-Bissau (6), the West African country with the highest prevalence of M. africanum Afri_1 strains (Technical Appendix Figure, panel B).
DESTUS Analysis
As indicated by cluster size analysis and RTIn–1, SIT 181 might be responsible for most recently transmitted TB cases in The Gambia. However, inferences about transmission that are purely based on cluster size analysis could be misleading because large clusters could equally be caused by an older strain that has been present for a long time or by strains that mutate slowly (13). To account for this imprecision, the DESTUS model was developed to factor the mutation rates of spoligotypes into analysis of genotypic clustering as a measure of emerging strains (13). DESTUS testing of our dataset for the whole MTBC or stratified by M. tuberculosis versus M. africanum found that SIT 181 was always detected as a highly significant emerging strain (p<10−30–10−31), followed by several other strains (Table 2). Similarly, SIT 181 was the only strain detected as emerging in the dataset from Guinea-Bissau (6).
Detecting Temporal Clusters of M. africanum SIT 181
To confirm previous results and to detect high rates of temporal clustering, we applied a purely temporal analysis to the M. africanum population by using the discrete Poisson model in SaTScan version 9.1.1 (www.satscan.org) (Table 3). We identified a significant (p = 0.001) temporal cluster of SIT 181 cases during August 2007–June 2008. This cluster showed an increased relative risk of 2.65 to the population for contracting SIT 181 when compared with the risk across the full study period (2002–2009).
Immunogenicity of MTBC Isolates
To identify associations between the emergence of certain genotypes and their immunogenicity, we used ELISPOT to measure PPD-induced interferon-γ responses in blood samples collected during 2002–2007 from patients infected with M. tuberculosis sensu stricto (n = 235), M. tuberculosis EA (n = 194), and M. africanum (n = 137). On the basis of the assumption that clustering is indicative of recent transmission, we compared PPD ELISPOT positivity of clustered strains with singletons (Table 4). Our data suggest that M. africanum strains clustered by spoligotyping are significantly more likely to produce a positive PPD ELISPOT result than are singletons (OR 31.78, 95% CI 9.24–109.28; p = 0.0001). For M. tuberculosis sensu stricto and M. tuberculosis EA, we found a similar, yet not significant, tendency.
After applying more stringent criteria than mere clustering, such as determining emerging strains (DESTUS), we compared PPD ELISPOT positivity of SIT 181 to singleton M. africanum Afri_1 strains; this analysis confirmed that SIT 181 is significantly more immunogenic than other types (OR 21.09, 95% CI 6.09–73.04; p = 0.0001). Similarly, we found a slight, not significant tendency for patients infected with SIT 181 to be more likely than patients infected with singleton strains to yield a positive Mantoux skin test (OR 1.15, 95% CI 0.30–4.44).
We found a correlation between MTBC strains of higher immunogenicity and their ability to spread within the human host population. In the study population in The Gambia, patients infected with strain SIT 181, the most prevalent M. africanum strain, were significantly more likely to yield a positive PPD ELISPOT result than were patients infected with M. africanum strains that do not have the ability to establish themselves within the human host population. To describe this association, we constructed a detailed population structure of MTBC isolates in which we analyzed 884 spoligotypes obtained from mycobacterial isolates from TB cases identified during 2002–2009. We found that most circulating strains belonged to either the EA or M. africanum Afri_1 clades, and therefore, we specifically focused on the comparison between these 2 lineages. On the basis of the assumption that clustered strains indicate recent transmission, we found that several M. tuberculosis strains appeared to have spread evenly within the host population. In contrast, most (59%) M. africanum transmission events and infections could be attributed to spoligotype SIT 181, which was responsible for 22% of all TB cases in the country. This result not only confirms recent findings from Guinea-Bissau, in which SIT 181 caused up to 49% of all M. africanum infections (6), but is also in agreement with a previous study of a smaller set of isolates from this study that showed a similar strain distribution within the 2 major lineages (Afri_1 and EA) (17).
Although it is widely accepted that genetic clusters are indicative of recent transmission, caution must be taken with the interpretation of such findings because the successful spread of a strain within the population—and thus genetic clustering—is highly dependent on 2 properties of the bacteria. For successful spread, strains must transmit from infected to uninfected host first; after this initial transmission, the infection must progress to active disease to be transmitted to the next susceptible host. However, only case–contact tracing studies, not molecular clustering data alone, can distinguish between transmission and progression of the different lineages. Consequently, we refer here to the spreading capacity of strains, rather than to transmission.
Our analysis using DESTUS (13), which correctly predicted the widely accepted emergence of Beijing lineage M. tuberculosis strains in other studies (13), detected SIT 181 as an unusually fast-growing strain relative to the mycobacterial background population of the sample. However, DESTUS does not take into account the migration history of Europeans and the mycobacteria they introduced into Africa. Because of this limitation, we sought to confirm our finding with a second approach and conducted a purely temporal analysis. We found that SIT 181 could have been emerging during a certain time in the study period, identifying a temporal cluster during 2007–2008 for which risk for infection with SIT 181 was 2.65-fold higher than that for the whole study period (2002–2009).
With SIT 181 constituting such a prominent cluster, it is conceivable that the strain’s high prevalence is related to selective pressure through, for instance, antimicrobial drug therapy. However, because resistance rates are relatively low in The Gambia (18), this explanation does not seem to apply. Therefore, we suggest another selective mechanism: we believe differences in spreading capacity and the interaction between M. tuberculosis and M. africanum populations might play a crucial role. In contrast to M. africanum, several clustered strains within the M. tuberculosis population have comparable potential to spread within the human host population, but no strain has a notable advantage over another, which results in a well-balanced population structure. We hypothesize that SIT 181, in its expanded ability to spread, resembles these M. tuberculosis strains more than it does strains with other spoligotype patterns in the M. africanum lineage. Thus, SIT 181 has a selective advantage and is able to compete with M. tuberculosis for the same biological niche within the human host.
Figure 2

Figure 2. . Linear regression analysis showing correlation between average quantitative purified protein derivative (PPD) response and recent transmission index (RTIn−1) for Mycobacterium tuberculosis complex isolates, The Gambia, 2002–2007. Open diamonds and dashed...
The nature and extent of epitope variation in M. tuberculosis strains is unclear. Findings range from highly variable T-cell epitopes within the esx gene family (19) to highly conserved epitopes when comparing genetic variation between predicted epitopes and the remainder of the M. tuberculosis genome (7). The latter study, which described T-cell epitopes as highly conserved, suggests that hyperconservation of T-cell epitopes is beneficial for the bacteria and could result in the successful spread of strains. In our study, we sought to understand whether the magnitude of an induced immune response was correlated with the spreading capacity of the bacteria, with a special focus on SIT 181. We therefore investigated PPD ELISPOT responses of patients infected with either lineage and found a nonsignificant tendency for clustered M. tuberculosis sensu stricto and EA strains toward being more likely than singletons to produce a positive PPD result. This small difference in immunogenicity might be in line with our hypothesis that the M. tuberculosis population spreads fairly homogenously. Consistent with the large observed differences in spreading capability, SIT 181 or clustered M. africanum strains have a 20- to 30-fold higher probability of yielding a positive PPD response (p<0.0001). This positive correlation between immunogenicity and spread becomes even more apparent when the RTI is plotted against the average quantitative PPD response (Figure 2). This comparison demonstrates not only the expected larger range in immunogenicity within the M. africanum lineage but also the phenotypic relatedness in immunogenicity of the highly spreading strains, independent of lineage.
An association between PPD response and spreading capacity is conceivable. A previous publication found that household contacts who slept in the same bedroom as an index case-patient (i.e., who were exposed to the highest infectious loads) had higher PPD ELISPOT responses than did less-exposed household contacts (20). However, further analysis will be needed to conclusively address whether spreading capacity is determined by infectious load; smear-positivity grade of cases; magnitude of the induced immune response; or an as-yet unknown immunogenic protein that enhances transmissibility, the absence of which (from strains of low spread) is merely reflected by a reduced ELISPOT response to PPD.
One possible limitation of spoligotype data is that the technique was designed on the basis of the clustered regularly interspaced short palindromic repeats (CRISPR) regions of M. tuberculosis sensu stricto and, therefore, could have a lower resolution when applied to M. africanum. Consequently, the large observed SIT 181 cluster could be a result of misclassification. Although the CRISPR regions of M. tuberculosis and M. africanum have not been extensively compared, we believe misclassification is very unlikely for 2 reasons. First, we found a comparable resolution of the technique for both lineages (18–19 genotypes/100 isolates). Second, when calculating the Hunter-Gaston Index (HGI), a measure for the discriminatory power of a technique, we found that HGI = 0.96 for spoligotyping of M. tuberculosis, HGI = 0.94 for M. africanum excluding SIT 181, and HGI = 0.64 for M. africanum with SIT181. Spoligotyping works equally well for 41% of M. africanum isolates and for M. tuberculosis isolates (0.94 vs. 0.96) but has drastically worse discriminatory power for the remaining 59% of M. africanum strains (0.64). A drop in HGI that was a result of misclassification within the M. africanum lineage could only result from a CRISPR region or mutation rate that was notably different between SIT 181 and the other M. africanum strains. However, this is unlikely because the remaining strains with HGI = 0.94 evolved out of SIT 181 and, thus, most likely have identical CRISPR regions and mutation rates. Therefore, by comparing these 3 HGI results, we can conclude that SIT 181 is a real cluster and not a result of misclassification. High-resolution genotyping methods, such as mycobacterial interspersed repetitive unit–variable number tandem repeat typing or whole-genome sequencing, is needed to conclusively confirm the genotypic homogeneity of the group of strains that constitute spoligotype pattern SIT 181.
We conclude that spoligotyping possesses comparable discriminatory resolution for M. tuberculosis and M. africanum. We were able to demonstrate that SIT 181 represents a strain (or family of strains) that clusters genotypically, temporally, and phenotypically and represents a major public health concern in West Africa, responsible for nearly one fourth of TB cases in The Gambia (22%) and Guinea-Bissau (23%) (6). Deciphering the virulence mechanisms that determine the differences in immunogenicity and spreading capacity between SIT 181 and the remaining singleton M. africanum strains will be key to improving TB prevention and transmission control in this region.
Dr Gehre is a postdoctoral researcher at the Institute for Tropical Medicine in Antwerp, Belgium, and seconded to the Medical Research Council, UK, The Gambia Unit. His current research focuses on TB, in particular, lineage-specific differences in virulence mechanisms and transmission dynamics of M. tuberculosis and M. africanum.
Acknowledgments
We thank Gunilla Källenius for approval to reanalyze the Guinea-Bissau dataset.
F.G. was supported by a “Travel Grant for a long stay abroad,” awarded by the Flemish Science Foundation.
References
- World Health Organization. Global tuberculosis control 2011. Geneva: The Organization; 2011 [cited 2012 Jul 1]. http://whqlibdoc.who.int/publications/2011/9789241564380_eng.pdf
- Castets M, Boisvert H, Grumbach F, Brunel M, Rist N. Tuberculosis bacilli of the African type: preliminary note [in French]. Rev Tuberc Pneumol (Paris). 1968;32:179–84 .PubMedGoogle Scholar
- de Jong BC, Antonio M, Gagneux S. Mycobacterium africanum—review of an important cause of human tuberculosis in West Africa. PLoS Negl Trop Dis. 2010;4:e744 and. DOIPubMedGoogle Scholar
- Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23 . DOIPubMedGoogle Scholar
- Comas I, Homolka S, Niemann S, Gagneux S. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS ONE. 2009;4:e7815 . DOIPubMedGoogle Scholar
- Groenheit R, Ghebremichael S, Svensson J, Rabna P, Colombatti R, Riccardi F, The Guinea-Bissau family of Mycobacterium tuberculosis complex revisited. PLoS ONE. 2011;6:e18601 . DOIPubMedGoogle Scholar
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42:498–503 . DOIPubMedGoogle Scholar
- van Soolingen D, de Haas PE, Hermans PW, Groenen PM, van Embden JD. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis. J Clin Microbiol. 1993;31:1987–95 .PubMedGoogle Scholar
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14 .PubMedGoogle Scholar
- Shabbeer A, Cowan LS, Ozcaglar C, Rastogi N, Vandenberg SL, Yener B, TB-Lineage: an online tool for classification and analysis of strains of Mycobacterium tuberculosis complex. Infect Genet Evol. 2012;12:789–97 . DOIPubMedGoogle Scholar
- Tang C, Reyes JF, Luciani F, Francis AR, Tanaka MM. spolTools: online utilities for analyzing spoligotypes of the Mycobacterium tuberculosis complex. Bioinformatics. 2008;24:2414–5 . DOIPubMedGoogle Scholar
- Reyes JF, Francis AR, Tanaka MM. Models of deletion for visualizing bacterial variation: an application to tuberculosis spoligotypes. BMC Bioinformatics. 2008;9:496 . DOIPubMedGoogle Scholar
- Tanaka MM, Francis AR. Detecting emerging strains of tuberculosis by using spoligotypes. Proc Natl Acad Sci U S A. 2006;103:15266–71 . DOIPubMedGoogle Scholar
- Kulldorff M. A spatial scan statistic. Commun Statist Theory Methods. 1997;26:1481–96.
- Hill PC, Brookes RH, Fox A, Fielding K, Jeffries DJ, Jackson-Sillah D, Large-scale evaluation of enzyme-linked immunospot assay and skin test for diagnosis of Mycobacterium tuberculosis infection against a gradient of exposure in The Gambia. Clin Infect Dis. 2004;38:966–73 . DOIPubMedGoogle Scholar
- Small PM, Hopewell PC, Singh SP, Paz A, Parsonnet J, Ruston DC, The epidemiology of tuberculosis in San Francisco. A population-based study using conventional and molecular methods. N Engl J Med. 1994;330:1703–9 . DOIPubMedGoogle Scholar
- de Jong BC, Antonio M, Awine T, Ogungbemi K, de Jong YP, Gagneux S, Use of spoligotyping and large sequence polymorphisms to study the population structure of the Mycobacterium tuberculosis complex in a cohort study of consecutive smear-positive tuberculosis cases in The Gambia. J Clin Microbiol. 2009;47:994–1001 . DOIPubMedGoogle Scholar
- Adegbola RA, Hill P, Baldeh I, Otu J, Sarr R, Sillah J, Surveillance of drug-resistant Mycobacterium tuberculosis in The Gambia. Int J Tuberc Lung Dis. 2003;7:390–3 .PubMedGoogle Scholar
- Uplekar S, Heym B, Friocourt V, Rougemont J, Cole ST. Comparative genomics of Esx genes from clinical isolates of Mycobacterium tuberculosis provides evidence for gene conversion and epitope variation. Infect Immun. 2011;79:4042–9 . DOIPubMedGoogle Scholar
- Hill PC, Fox A, Jeffries DJ, Jackson-Sillah D, Lugos MD, Owiafe PK, Quantitative T cell assay reflects infectious load of Mycobacterium tuberculosis in an endemic case contact model. Clin Infect Dis. 2005;40:273–8 . DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleReference has only first page number. Please provide the last page number if article is longer than one page. (in reference 4 "Brudey, Driscoll, Rigouts, Prodinger, Gori, Al-Hajoj, et al., 2006").
Reference has only first page number. Please provide the last page number if article is longer than one page. (in reference 12 "Reyes, Francis, Tanaka, 2008").
Table of Contents – Volume 19, Number 10—October 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Florian Gehre, Mycobacteria Unit, Institute for Tropical Medicine (ITM), Nationalestraat 155, 2000 Antwerp, Belgium
Top