Volume 19, Number 5—May 2013
Research
Full-Genome Deep Sequencing and Phylogenetic Analysis of Novel Human Betacoronavirus
Matthew Cotten, Tommy T. Lam, Simon J. Watson, Anne L. Palser, Velislava Petrova, Paul Grant, Oliver G. Pybus, Andrew Rambaut, Yi Guan, Deenan Pillay, Paul Kellam , and Eleni Nastouli
, and Eleni Nastouli
Figure 4
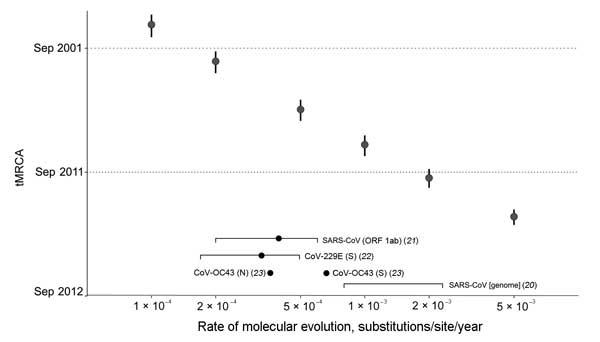
Figure 4. . . . tMRCA analysis across a range of fixed evolutionary rates. The tMRCA of EMC/2012 and England/Qatar/2012 estimated from fixing a range of genomic evolutionary rates (1 × 10−4, 2 × 10−4, 5 × 10−4, 1 × 10−3, 2 × 10−3, and 5 × 10−3 substitutions/site/year) are shown in data points with vertical error bars (95% highest posterior density). Evolutionary rate estimates of human CoV genome and genes in the literature are indicated at the bottom of the plot (mean or point estimate as a dot, 95% CIs of estimate as a square bracket). tMRCA, time to the most recent common ancestor; CoV, coronavirus; SARS, severe acute respiratory syndrome; ORF, open reading frame.
Page created: April 23, 2013
Page updated: April 23, 2013
Page reviewed: April 23, 2013
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.