Volume 19, Number 9—September 2013
Research
Continued Evolution of West Nile Virus, Houston, Texas, USA, 2002–2012
Cite This Article
Citation for Media
Abstract
We investigated the genetics and evolution of West Nile virus (WNV) since initial detection in the United States in 1999 on the basis of continual surveillance studies in the Houston, Texas, USA, metropolitan area (Harris County) as a surrogate model for WNV evolution on a national scale. Full-length genomic sequencing of 14 novel 2010–2012 WNV isolates collected from resident birds in Harris County demonstrates emergence of 4 independent genetic groups distinct from historical strains circulating in the greater Houston region since 2002. Phylogenetic and geospatial analyses of the 2012 WNV isolates indicate closer genetic relationship with 2003–2006 Harris County isolates than more recent 2007–2011 isolates. Inferred monophyletic relationships of these groups with several 2006–2009 northeastern US isolates supports potential introduction of a novel WNV strain in Texas since 2010. These results emphasize the need to maintain WNV surveillance activities to better understand WNV transmission dynamics in the United States.
The emergence of West Nile virus (WNV) in the Western Hemisphere in 1999 poses an ongoing public health threat in North America as the most common cause of epidemic encephalitis in the United States (1). WNV transmission is maintained in an enzootic cycle between mosquitoes and birds; equids, humans, other mammals, and some bird species act as dead-end hosts (2). Human infections are asymptomatic in 80% of cases, and West Nile fever develops in ≈20% of infected patients, which progresses to neuroinvasive disease in <1% (3).
After introduction of WNV in the United States in 1999 (4), local transmission of the original New York genotype (NY99) in resident Culex spp. mosquito and wild bird populations fueled the geographic expansion of WNV from the northeastern region across the continental United States, north into Canada, and south into Central and South America (5–7). Subsequent introduction into Texas in 2002 resulted in 105 confirmed human infections, high mortality rates among local corvids, and a 31.2% seroconversion rate among resident birds of Harris County, Texas (Houston metropolitan area) alone (8). Uninterrupted surveillance of the related St. Louis encephalitis virus in local Culex spp. mosquito populations by the Harris County Mosquito Control Division since 1964 provided an ideal infrastructure for the expanded detection of WNV activity in the mosquito vector and the wild bird reservoir on a major bird migratory pathway. Routine collections of WNV-positive birds and mosquito pools to date have provided an outstanding opportunity to investigate WNV diversity and evolution on a fine geographic scale comparable to similar surveillance foci in the midwestern and New England regions of the United States (9–11). However, because of its geographic location, Harris County represents a different ecosystem, namely a warm year-round climate with unique resident mosquito and avian species.
Phylogenetic examination of 2002–2004 Harris County isolates confirmed rapid displacement of the NY99 genotype with the novel North American genotype (NA/WN02) in 2002 (12). Fine-scale geospatial genetic comparisons of these isolates provided further evidence of increased WNV genetic diversification in the greater Houston region relative to the homogenous distribution of the now extinct NY99 genotype (12,13). Subsequently, McMullen et al. identified the emergence of the southwestern genotype (SW/WN03) in the southwestern United States in 2003 and positive selection for the encoded NS4A-A85T and NS5-K314R amino acid substitutions in the WNV nonstructural (NS) proteins (14). To date, the NA/WN02 and SW/WN03 genotypes still appear to co-circulate.
Endemic transmission of WNV in the United States since 2006 has shown a dramatic decrease in the confirmed incidence of clinical WNV disease; <1,100 annual human cases were reported during 2008–2011 (15). Despite identification of regional heterogeneous WNV populations, a relative stasis in WNV evolution has been observed in Harris County, consistent with the logistic molecular clock model and a decreasing viral growth rate proposed on a national scale (16–18). Notably, the current 2012 WNV transmission season demonstrates major divergence from this status quo; >5,600 human infections have been reported nationwide (15). Incidence of clinical WNV disease in the Texas outbreak alone accounted for >33% of the cases in the United States (1,868 cases, including 844 reports of neuroinvasive disease and 89 deaths) and >994 confirmed cases in the greater Dallas/Fort Worth, Texas, metropolitan area and 101 cases in Harris County (15,19). These changes reflect final US and Texas and WNV cases for 2012 reported by the Centers for Disease Control and Prevention (Atlanta, GA, USA) (www.cdc.gov/media/releases/2013/a0513-west-nile.html) after submission of this report after review. Therefore, studies concerning the continued evolution of WNV in the central and southern United States remain vital for elucidating the role of dynamic genetic heterogeneity and accumulation of novel mutations in the transmission dynamics and incidence of clinical WNV disease.
We report consensus sequence analyses of 17 novel full-length 2010 (n = 1), 2011 (n = 1), and 2012 (n = 15) WNV isolates collected from WNV-positive birds and Culex spp. mosquito pools in Harris County Texas (n = 14) and the greater Dallas/Fort Worth region (n = 3). Inclusion of these new isolates with 28 additional 2002–2009 Harris County WNV isolates in phylogenetic and geospatial analyses of the greater Houston region provides an ideal model for investigating the role of ongoing WNV evolution relative to environmental and clinical incidence reported over the past decade. Furthermore, isolates from the recent WNV epidemic demonstrate closer phylogenetic relationships with original 2002–2003 Harris County isolates, inconsistent with phylogenetic trends observed until 2011 and supporting evidence for the recent introduction of a novel WNV strain(s) in Texas from another geographic region.
Virus Isolates
The World Reference Center for Emerging Viruses and Arboviruses at the University of Texas Medical Branch in Galveston, Texas, supplied the WNV isolates characterized herein provided by the Harris County Mosquito Control Division and Texas Department of State Health Services (Table 1). The fourteen 2010–2012 Harris County isolates were cultured from brain tissue of collected dead WNV-positive birds in Vero cells at the University of Texas Medical Branch in Galveston. Secondary passage of each isolate in Vero cells provided working stocks that were stored at –80°C. These same procedures were used for the 3 Dallas/Fort Worth mosquito isolates.
Genomic Sequencing
Extraction of viral RNA, reverse transcription PCR, and sequencing was conducted according to established protocols (12,14). Resulting sequences were aligned and edited relative to the prototype NY99-flamingo382-99 strain (AF196835) by using ContigExpress in the VectorNTI program (Invitrogen, Carlsbad, CA, USA) (4). Sequences were assembled in BioEdit version 7.0.9.0 (20) with 358 full-length North American WNV isolates published in GenBank (as of November 2012) and the IS-98 STD (AF481864) Israeli isolate (21). The encoded 10,299 nt open reading frames (ORFs) for all 372 WNV isolates were aligned by using the MUSCLE algorithm (22); the ORF was used because some viruses published in GenBank include the ORF alone and not the entire genome. Two additional alignments included the ORF of all 42 2002–2012 Harris County WNV isolates with and without the three 2012 Dallas/Fort Worth isolates and the prototype NY99-flamingo382-99 and IS-98 STD reference strains. Screening for potential site-specific positive selection was performed by using the Datamonkey server (23,24) and the single-likelihood ancestor counting, fixed effect likelihood (FEL), and internal branches FEL methods (25,26). Positive selection was defined as dN>dS and a p value <0.05 in >1 method.
Phylogenetic Analyses
Neighbor-joining (NJ) phylogenetic analyses of all alignments were processed in Seaview version 4.3.0 by using the Hasegawa–Kishino–Yano 85 substitution model and 10,000 bootstrap replicates (27). Maximum-likelihood (ML) analyses used RAxML-HPC Blackbox version 7.3.2 (28,29) on the CIPRES Science Gateway version 3.1 server (30) with the generalized time reversible substitution model with invariable sites, a gamma distribution, and 1,000 bootstrap replicates. Bayesian-inferred coalescent phylogenies were produced in BEAST version 1.6.2 (http://beast.bio.ed.ac.uk) by using the generalized time reversible substitution model with invariable sites and a gamma distribution with applied taxa dates, an uncorrelated lognormal relaxed clock model, and Bayesian Skyline prior constraints (31). Resulting BEAST.log and TRE files were down-sampled from triplicate 50,000,000 state runs by using LogCombiner version 1.7.4 and validation in Tracer version 1.5 (31). Inferred phylogenetic trees were edited in FigTree version 1.3.1 (www.mybiosoftware.com/phylogenetic-analysis/2407). In all NJ and ML tree topologies, the IS-98 STD isolate was used as a common phylogenetic outgroup.
Statistical Analyses
The Fischer exact test and post hoc analyses were performed at α = 0.05 (IBM SPSS statistics version 20; IBM, Armonk, NY, USA) to test the association between year of collection and determined phylogenetic groupings. Adjusted standardized residuals (z-scores) at α = 0.05 were compared against the critical z-value (± 1.96) with a Bonferroni correction for multiple comparisons.
WNV Collection
A total of 14 WNV isolates from Harris County (Table 1) were examined. Two isolates were collected from house sparrows (Passer domesticus) in 2010 and 2011, and 12 isolates were obtained from WNV-positive birds in 2012: 10 from blue jays (Cyanocitta cristata) and 1 each from a loggerhead shrike (Lanius ludovicianus) and a house sparrow.
Phylogenetic and geospatial analyses characterized herein compared all 14 WNV isolates with 28 other published Harris County isolates collected during 2002–2009 from mosquito pools (n = 12), birds (n = 13), or humans (n = 3) (12,14,16). Genomic sequences were determined for WNV isolates from each year except in 2004: 2002 (n = 3), 2003 (n = 8), 2005 (n = 2), 2006 (n = 3), 2007 (n = 4), 2008 (n = 1), and 2009 (n = 7). Sample coverage was restricted on the basis of sequence and sample availability. The Kuritz (also known as TVP8533; GenBank accession no. AY289214) isolate (Southeast Coastal Texas genotype) was included as a common Texas outgroup, and the TX114 (Bird114; GU827998) isolate served as the prototypic member of the NA/WN02 genotype (12,32).
Divergent WNV Evolution
Nucleotide Changes
Comparison of the 14 novel Harris County isolates with the NY99 (NY99-flamingo382-99) prototype strain identified 45–79 nt differences (0.41%–0.72%) per 11,029-nt genome (4). Each isolate encodes 8 of the 13 nt changes characteristic of the NA/WN02 genotype (12). In addition, novel U→C transitions at positions 7015 in the NS4B gene and 8811 in the NS5 gene were conserved in 11 of the 12 Harris County isolates from 2012 (except TX8604).
Amino Acid Substitutions
These 14 isolates differed at 48 unique residues in the encoded 3,433-aa polyprotein relative to NY99 and had 2–10 (0.06%–0.29%) substitutions per isolate. Each isolate encoded the E-V159A substitution characteristic of the NA/WN02 genotype (12); however, the signature NS4A-A85T SW/WN03 genotype substitution (14) was identified only in the 2011 TX8349 isolate. Furthermore, 11 of the 48 deduced amino acid substitutions were conserved in >1 isolate. Single-likelihood ancestor counting, FEL, and internal branches FEL method analysis identified potential positive selection of the NS2A-H119Y substitution in the 2012 TX8546 isolate.
Phylogenetic and Geospatial Analysis
Figure 1
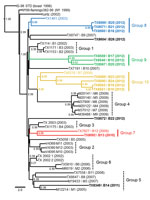
Figure 1. . Bayesian-inferred, 50% majority-rule, coalescent phylogenetic tree of published, full-length West Nile virus isolates, Harris County, Texas, USA, 2002–2012. Novel 2010–2012 Harris County isolates cluster into 4 distinct monophyletic groups designated...
Phylogenetic reconstruction of ancestral topologies among all 42 published 2002–2012 Harris County WNV isolates used NJ, ML, and relaxed clock Bayesian coalescent methods. Consistent tree topologies rooted to the common IS-98 STD isolate outgroup were produced with all 3 methods. Inclusion of 2010–2012 isolates retained the phylogenetic clustering of the 2002–2009 Harris County isolates within the 6 monophyletic groups (groups 1-6) proposed by McMullen et al. (14) (Figure 1). Conserved nucleotide and amino acid divergence among the fourteen 2010–2012 isolates indicates emergence of 4 novel monophyletic clusters of avian isolates (groups 7–10) (Table 2). Furthermore, groups 7–10 demonstrate a significant (p≤0.044) relationship between year of collection and identified monophyletic lineage with more 2012 isolates clustering within groups 8–10.
Group 7 (0.57% nt divergence from NY99) includes the 2009 TX7827 and 2010 TX8092 isolates. Group 8 consists of the 2003 TX1461 (Bird1461) outgroup and three 2012 isolates: TX8560, TX8571, and TX8599 (0.01%–0.04%). Group 9 (0.40%–0.45%) includes three 2012 isolates: TX8546, TX8559, and TX8567. Group 10 includes the 2006 TX6276 isolate and 2012 isolates from this study: TX8551, TX8562, TX8589, and TX8590 (0.00%–0.61%). Overall, the 37 Harris County isolates in groups 1–10 differ at 80 nt positions and 27 had substitutions shared in >1 isolate (Technical Appendix). Five substitutions (prM-V156I, NS2A-M90V, NS2A-L95F, NS4B-I240M, and NS4B-E249G) were conserved in >1 phylogenetic group. Furthermore, the signature NS4A-A85T SW/WN03 genotype substitution was present only in the 2011 TX8349 and other group 5 isolates. The TX8572 and TX8604 isolates cluster as outliers (>0.44% nt divergence) to the proposed phylogenetic groups and show increased divergence (1.13% nt and 0.38% aa) between these 2 isolates.
Figure 2
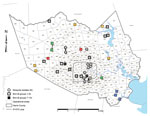
Figure 2. . Incidence of vector-borne West Nile Virus (WNV) in Harris County, Texas, USA, 2002–2012, showing cumulative distribution of confirmed avian and mosquito (Culex and Aedes spp.) WNV isolates. Small numbers indicate...
Superimposition of 38 of the 42 Harris County isolates (4 isolates were excluded because of unknown collection location) on a vector-borne WNV incidence map of Harris County, based on known collection information in the 268 mosquito control operational areas, highlights the shift in WNV circulation patterns across the greater Houston region over the past decade (Figure 2). Mosquito pool isolates demonstrate robust genetic homogeneity and limited geographic distribution (Figure 2) in 9 of the 268 operational areas, suggestive of WNV overseasoning in resident mosquito populations. In particular, isolates M1 (2005) and M3 (2007) cluster within group 5, indicating transmission of the same virus strain in Houston across multiple years. In contrast, avian-derived isolates illustrate widespread incidence of similar genetic signatures in 2002–2011 WNV isolates, as highlighted in dispersal of several 2005–2011 group 5 SW/WN03 genotype isolates. However, group 7–10 isolates demonstrate comparable geographic distribution but limited monophyletic support with group 1–6 isolates. Furthermore, group 8 isolates remain geographically restricted compared to more pervasive group 10 isolates. Overall, phylogenetic and geospatial analysis of novel 2012 WNV isolates indicate a closer genetic relationship with 2003–2006 Harris County isolates than with more recent 2007–2011 WNV isolates.
Novel Introduction Event in 2012 WNV Outbreak in Texas
Figure 3

Figure 3. . Evolution of West Nile virus (WNV) in North America, 1999–2012. A) Bayesian coalescent tree of all published North American WNV isolates. The NY99 (NY99) and southwestern (SW/WN03) genotypes flank the...
Inclusion of the fourteen 2010–2012 Harris County isolates in an additional phylogenetic analysis with 358 published North American WNV isolates enabled us to evaluate the potential influence of active WNV transmission in North America on the recent WNV evolution dynamics observed in Harris County. NJ, ML, and Bayesian relaxed clock methods produced consistent overall tree topologies with retention of the published NY99, NA/WN02 (12), and SW/WN03 (14) genotypes (Figure 3, panel A). Groups 7–10 showed conserved clustering within the NA/WN02 genotype and robust (≥0.90 posterior probabilities) monophyletic support for shared lineage with several 2006–2009 New York and Connecticut isolates (Figure 3, panels B–F). Furthermore, the 2010 TX8092 isolate demonstrated consistent monophyletic clustering within the NA/WN02 genotype with the 2009 TX7827 and additional 2008 New York (WNV-1/US/BID-v4622/2008) isolates (Figure 3, panel G). In contrast, the novel 2011 TX8349 Harris County isolate retained topologic distribution within the SW/WN03 genotype. Outside the identified group 7–10 monophyletic clusters, all inferred basal node topologies exhibited poor statistical support (≤0.70 posterior probabilities) within the NA/WN02 genotype.
Principal support for the identified monophyletic lineages is based on limited nucleotide divergence (<0.65%) and retention of unique substitutions between the 2010 and 2012 Harris County isolates and those from the northeastern United States compared with published isolates from Texas and the southwestern United States. In particular, the group 9 monophyletic lineage (Figure 3, panel E) encodes several conserved substitutions: NS2A-T52I, NS2A-L95F, NS3-S334T, and NS4B-S14I with a single NS2A-H119Y substitution shared between the 2008 New York (WNV-1/US/BID/v4097/2008) outgroup and the 2012 TX8546 isolate. Each isolate also encodes the characteristic E-V159A substitution with the conserved absence of the NS4A-A85T and NS5-K314R substitutions supporting monophyletic distribution and ancestral lineage within the NA/WN02 versus NY99 and SW/WN03 genotypes.
Harris County Paradigm—Model for WNV Evolution
Figure 4
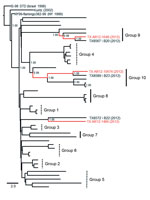
Figure 4. . Phylogenetic support for expanded application of the proposed Harris County, Texas, USA, paradigm as a model for West Nile virus (WNV) evolution during the Dallas/Fort Worth, Texas, outbreak, 2012. Bayesian...
To evaluate application of the proposed Harris County paradigm outside the greater Houston metropolitan region, we collected 3 WNV isolates from Culex spp. mosquito pools in the recent 2012 Dallas/Fort Worth, Texas WNV outbreak (Table 1) were sequenced and included in a Bayesian phylogenetic analysis with all 42 2002–2012 Harris County isolates. Both Collin County isolates (TX AR12-1648 and TX AR12-10674) exhibited limited nucleotide divergence (≤0.30%) and robust monophyletic clustering (≥0.98 posterior probabilities) with several group 9 and 10 isolates in the Harris County paradigm (Figure 4). In addition, the TX AR12-1468 Denton County isolate demonstrated shared lineage with the nonclustering TX8572 isolate. Overall, our results support potential application of the Harris County paradigm as a relevant model for WNV evolution in Texas as a whole and, possibly, on a national scale.
Surveillance of WNV transmission in Harris County, Texas, provides an excellent model for elucidating the dynamics of endemic and epidemic WNV evolution on a fine geographic scale. The southeastern coastal region of Texas serves as a temporary roosting site on a major flyway for migratory birds in transit between more temperate and tropical regions of the Americas. Consequently, in addition to resident bird populations, this region hosts more avian species (and possible WNV reservoir hosts) than anywhere else in the United States. Prior applications of the proposed Harris County paradigm confirmed emergence of NA/WN02 (12) and SW/WN03 (14) genotypes from the United States in 2002 and 2003, respectively, as a surrogate model for WNV evolution on a national scale. Our analyses support emergence of 4 novel monophyletic groups of 2010–2012 Harris County isolates (groups 7–10) distinct from group 1–6 phylogenetic clusters identified (Figure 1) (14). Furthermore, these isolates exhibit closer ancestral lineage with 2002–2003 Harris County isolates compared with more recent 2007–2009 strains. The single 2011 TX8349 isolate clusters with several 2005–2008 isolates within the group 5 SW/WN03 genotype with conserved expression of the signature NS4A-A85T substitution.
Restriction of conserved substitutions to individual monophyletic groups (Table 2) indicates increased genetic heterogeneity among circulating 2010–2012 populations than with earlier 2002–2009 Harris County WNV populations. Geographic reconstruction of the 42 WNV isolates on a map of Harris County based on mosquito control operational areas (Figure 2) supports a heterogeneous transmission model with limited correlation between fine-scale geographic dispersion and sequence divergence over time (9,10). However, regional phylogenetic foci among group 2, 4, and 5 mosquito pools and group 8 avian isolates supports potential homogeneous and trans-seasonal WNV transmission in localized vector populations as documented in other studies (11,13,33).
Before the 2010–2012 phylogenetic analyses, a relative stasis in WNV evolution had been observed in Harris County (16) and the northeastern United States (11,13,17,33) after confirmed emergence of the NA/WN02 and SW/WN03 genotypes. Fine-scale geographic phylogenetic analyses in suburban Chicago and Illinois have identified maintenance and active evolution of heterogeneous WNV populations in local mosquito and avian vectors from the initial introduction into the United States in 1999 until 2008 but not more recently (9,10,18). Applied genomic and phylogenetic comparisons highlight the major divergence of the fourteen 2010–2012 Harris County isolates from historical dynamics of local WNV evolution and emergence of 3 distinct monophyletic lineages (groups 8–10) in 2012 (Figure 1). However, the 2010 and 2012 Harris County isolates exhibit major sequence divergence (>0.42%) relative to group 1–6 isolates despite shared geographic distribution and environmental conditions. In addition, co-circulation of these novel genetic signatures was confirmed in 3 Culex spp. mosquito pool isolates obtained from the 2012 WNV outbreak in the greater Dallas/Fort Worth region. On the basis of these observations, introduction of a novel or existing strain from the United States into the circulating greater Houston WNV populations, and possibly Texas as a whole, since 2010 offers an alternative explanation for these divergent genetic signatures. However, our results do not exclude possible emergence of related existent viral populations as dominant regional strains in the recent epidemic WNV transmission season.
To test these hypotheses, we included fourteen 2010–2012 Harris County isolates in a comprehensive Bayesian phylogenetic analysis with all 358 published North American WNV isolates. Shared lineage of group 7–10 isolates with several 2006–2009 strains form the northeastern United States within the NA/WN02 genotype was identified with robust monophyletic support (≥0.90 posterior probabilities) (Figure 3, panels B–G). Principal evidence for these monophyletic lineages is highlighted on the basis of limited divergence (≤0.65%) and conserved expression of unique substitutions relative to observed genetic diversity (0.8%–1.0%) between published group 1–6 strains from the southwestern United States (14).
Our results support inferred lineage of 2010 and 2012 Harris County isolates from ancestral strains in the northeastern United States, consistent with a single or multiple introduction event(s) during 2010–2012 in the greater Houston region. However, poor statistical confidence (≤0.70 posterior probabilities) for all inferred basal node topologies within the NA/WN02 genotype limits direct comparison of independent evolution between Harris County isolates in different monophyletic lineages. Furthermore, consistent phylogenetic grouping of the 2011 TX8349 isolate within the SW/WN03 genotype indicates co-circulation of this genotype in Harris County until the 2012 transmission season. However, limited clustering of sequenced 2010–2012 Harris County isolates within the SW/WN03 genotype may be an artifact of inherent bias in sample collection.
Sampling bias is a recognized constraint in phylogenetic and paired geospatial analyses. Unfortunately, the predominance of WNV surveillance remains restricted to limited regional foci. Of the 372 full-length WNV isolates in our analyses, 31.5% (n = 117) were collected in Connecticut, 22.0% (n = 82) in New York, 16.4% (n = 61) in Texas, and 8.1% (n = 30) in Illinois during 1999–2012. In contrast, a single isolate has been characterized for each of the midwestern/central states of Michigan, North Dakota, and South Dakota, all of which reported a major increase in the number of clinical WNV cases (n≥89) in the 2012 epidemic season not seen since 2007 (15). Increased WNV surveillance in mosquito and avian vector populations across the entire United States is needed to provide critical, unbiased insight into the underlying dynamics of WNV evolution and transmission in US host populations.
The Harris County Public Health and Environmental Services Mosquito Control Division, which was founded in response to a St. Louis encephalitis virus outbreak in 1964, provides a model infrastructure for WNV surveillance in resident mosquito and avian populations in the greater Houston region (8). Uninterrupted collection and processing of WNV-positive bird and mosquito pools since the 2002 introduction of WNV into Harris County has provided a conduit for the scientific investigation of real-time disease outbreaks with direct translation of findings towards optimized vector-borne disease control and prevention. Incorporation of this paradigm in public health directives across the United States would provide a proactive approach towards detection and response to clinical outbreak scenarios of endemic and exotic pathogens in the United States.
In conclusion, retrospective analysis of WNV evolution in Harris County over the past decade indicates a recent shift in the genetic and phylogenetic signature of circulating WNV populations, designated the Harris County paradigm. Further evidence supports introduction of a strain into Texas from the northeastern United States since 2010. Continued WNV surveillance is needed to confirm the effect of this genetic shift in the transmission dynamics and incidence of clinical WNV disease in the greater Houston and surrounding US regions.
Mr Mann is a predoctoral student at the University of Texas Medical Branch in Galveston, Texas. His research interests include the pathogenesis and molecular epidemiology of flaviviruses.
Acknowledgments
We thank the Texas Department of State Health Services Laboratory Arbovirus Team for generously providing WNV samples from the Dallas/Fort Worth region, and Amy Schuh for providing helpful discussions and statistical insight in preparation of this manuscript.
This study was supported in part by National Institutes of Health grant AI 067847 to A.D.T.B. and contract HHSN272201000040I/HHSN27200004/D04 to R.B.T. B.R.M. and A.R.M. were supported by National Institutes of Health T32 training grant AI 007526 from the National Institute of Allergy and Infectious Diseases.
References
- Davis LE, Debiasi R, Goade DE, Haaland KY, Harrington JA, Harnar JB, West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286–300. DOIPubMedGoogle Scholar
- Blitvich BJ. Transmission dynamics and changing epidemiology of West Nile virus. Anim Health Res Rev. 2008;9:71–86. DOIPubMedGoogle Scholar
- Hayes EB, Gubler DJ. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu Rev Med. 2006;57:181–94. DOIPubMedGoogle Scholar
- Lanciotti RS, Roehrig JT, Deubel V, Smith J, Parker M, Steele K, Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286:2333–7. DOIPubMedGoogle Scholar
- Pepperell C, Rau N, Krajden S, Kern R, Humar A, Mederski B, West Nile virus infection in 2002: morbidity and mortality among patients admitted to hospital in southcentral Ontario. CMAJ. 2003;168:1399–405 .PubMedGoogle Scholar
- Deardorff E, Estrada-Franco JG, Brault AC, Navarro-Lopez R, Campomanes-Cortes A, Paz-Ramirez P, Introduction of West Nile virus strains in Mexico. Emerg Infect Dis. 2006;12:314–8. DOIPubMedGoogle Scholar
- Morales MA, Barrandeguy M, Fabbri C, Garcia JB, Vissani A, Trono K, West Nile virus isolation from equines in Argentina, 2006. Emerg Infect Dis. 2006;12:1559–61. DOIPubMedGoogle Scholar
- Lillibridge KM, Parsons R, Randle Y, Travassos da Rosa AP, Guzman H, Siirin M, The 2002 introduction of West Nile virus into Harris County, Texas, an area historically endemic for St. Louis encephalitis. Am J Trop Med Hyg. 2004;70:676–81 .PubMedGoogle Scholar
- Bertolotti L, Kitron U, Goldberg TL. Diversity and evolution of West Nile virus in Illinois and the United States, 2002–2005. Virology. 2007;360:143–9 . DOIPubMedGoogle Scholar
- Bertolotti L, Kitron UD, Walker ED, Ruiz MO, Brawn JD, Loss SR, Fine-scale genetic variation and evolution of West Nile virus in a transmission “hot spot” in suburban Chicago, USA. Virology. 2008;374:381–9. DOIPubMedGoogle Scholar
- Armstrong PM, Vossbrinck CR, Andreadis TG, Anderson JF, Pesko KN, Newman RM, Molecular evolution of West Nile virus in northern temperate region: Connecticut, USA 1999–2008. Virology. 2011;417:203–10. DOIPubMedGoogle Scholar
- Davis CT, Ebel GD, Lanciotti RS, Brault AC, Guzman H, Siirin M, Phylogenetic analysis of North American West Nile virus isolates, 2001–2004: evidence for the emergence of a dominant genotype. Virology. 2005;342:252–65. DOIPubMedGoogle Scholar
- Ebel GD, Dupuis AP, Ngo K, Nicholas D, Kauffman E, Jones SA, Partial genetic characterization of West Nile virus strains, New York State, 2000. Emerg Infect Dis. 2001;7:650–3 .PubMedGoogle Scholar
- McMullen AR, May FJ, Li L, Guzman H, Bueno R Jr, Dennett JA, Evolution of new genotype of West Nile virus in North America. Emerg Infect Dis. 2011;17:785–93. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention (CDC). West Nile virus: statistics, surveillance, and control archive, 1999–present [cited 2012 Jun 18]. http://www.cdc.gov/ncidod/dvbid/westnile/surv&control_archive.htm
- Davis CT, Li L, May FJ, Bueno R Jr, Dennett JA, Bala AA, Genetic stasis of dominant West Nile virus genotype, Houston, Texas. Emerg Infect Dis. 2007;13:601–4. DOIPubMedGoogle Scholar
- Snapinn KW, Holmes EC, Young DS, Bernard KA, Kramer LD, Ebel GD. Declining growth rate of West Nile virus in North America. J Virol. 2007;81:2531–4. DOIPubMedGoogle Scholar
- Amore G, Bertolotti L, Hamer GL, Kitron UD, Walker ED, Ruiz MO, Mutli-year evolutionary dynamics of West Nile virus in suburban Chicago, USA, 2005–2007. Phil Trans R Soc B. 2010;365:1871–8.
- Texas Department of State Health Services (DSHS). West Nile virus in Texas 2012–present [cited 2012 Jun 18]. http://www.dshs.state.tx.us/idcu/disease/arboviral/westNile/
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series. 1999;41:95–8.
- Malkinson M, Banet C, Weisman Y, Pokamunski S, King R, Drouet MT, Introduction of West Nile virus in the Middle East by migrating white storks. Emerg Infect Dis. 2002;8:392–7. DOIPubMedGoogle Scholar
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. DOIPubMedGoogle Scholar
- Delport W, Poon AF, Frost SD, Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26:2455–7. DOIPubMedGoogle Scholar
- Pond SL, Frost SD. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21:2531–3. DOIPubMedGoogle Scholar
- Kosakovsky Pond SL, Frost SD. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–22. DOIPubMedGoogle Scholar
- Pond SL, Frost SD, Grossman Z, Gravenor MB, Richman DD, Brown AJ. Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLOS Comput Biol. 2006;2:e62. DOIPubMedGoogle Scholar
- Gouy M, Guidon S, Gascuel O. Seaview version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–4. DOIPubMedGoogle Scholar
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. DOIPubMedGoogle Scholar
- Stamatakis A, Hoover P, Rougemont J. A fast bootstrapping algorithm for the RAxML web-servers. Syst Biol. 2008;57:758–71. DOIPubMedGoogle Scholar
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE); November 14, 2010; New Orleans, Louisiana. Red Hook (NY): Curran Associates; 2010. p. 1–8.
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214 . DOIPubMedGoogle Scholar
- Granwehr BP, Li L, Davis CT, Beasley DW, Barrett AD. Characterization of a West Nile virus isolate from a human on the Gulf Coast of Texas. J Clin Microbiol. 2004;42:5375–7 . DOIPubMedGoogle Scholar
- Ebel GD, Carricaburu J, Young D, Bernard KA, Kramer LD. Genetic and phenotypic variation of West Nile virus in New York, 2000–2003. Am J Trop Med Hyg. 2004;71:493–500 .PubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 19, Number 9—September 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Alan D.T. Barrett, University of Texas Medical Branch, 3.230 Mary Moody Northen Pavilion, 301 University Blvd, Galveston, TX 77555-0436, USA
Top