Volume 20, Number 4—April 2014
Research
High Rates of Antimicrobial Drug Resistance Gene Acquisition after International Travel, the Netherlands
Cite This Article
Citation for Media
Abstract
We investigated the effect of international travel on the gut resistome of 122 healthy travelers from the Netherlands by using a targeted metagenomic approach. Our results confirm high acquisition rates of the extended-spectrum β-lactamase encoding gene blaCTX-M, documenting a rise in prevalence from 9.0% before travel to 33.6% after travel (p<0.001). The prevalence of quinolone resistance encoding genes qnrB and qnrS increased from 6.6% and 8.2% before travel to 36.9% and 55.7% after travel, respectively (both p<0.001). Travel to Southeast Asia and the Indian subcontinent was associated with the highest acquisition rates of qnrS and both blaCTX-M and qnrS, respectively. Investigation of the associations between the acquisitions of the blaCTX-M and qnr genes showed that acquisition of a blaCTX-M gene was not associated with that of a qnrB (p = 0.305) or qnrS (p = 0.080) gene. These findings support the increasing evidence that travelers contribute to the spread of antimicrobial drug resistance.
Antimicrobial drug resistance is a public health threat worldwide that limits clinical treatment options for bacterial infections. Most research on antimicrobial drug resistance has been focused on resistance in clinically relevant pathogenic bacteria. However, a vast and largely unexplored reservoir of resistance genes is present in nonpathogenic bacteria living in the environment or as commensal agents (1–5). Because of horizontal gene transfer (HGT) among microbes of diverse species and genera, antimicrobial drug resistance mechanisms in an organism, regardless of whether it is a pathogen, have the potential to emerge in clinically relevant pathogens (6). Several of such HGT interactions between clinically relevant pathogens and environmental species have been described; for example, the plasmid-mediated quinolone resistance encoding qnrA gene originated from the chromosomes of the aquatic bacterium Shewanella algae (7). Another well-known example is the extended-spectrum β-lactamase (ESBL) encoding blaCTX-M gene, which originates from chromosomal genes of environmental Kluyvera species (8) and has emerged as the most prevalent cause of plasmid-mediated ESBL.
Resistance reservoirs have unpredictable and immense potential for rendering antimicrobial drugs ineffective. The human gut microbiota warrants special attention because of its high density of microorganisms and high accessibility (9). The gastrointestinal tract is constantly exposed to numerous bacteria from the environment, e.g., food, water, soil, other humans, or animals. These incoming bacteria often harbor antimicrobial drug resistance genes (10), which can be transferred to the indigenous microbial communities through HGT, where they may enrich the pool of available antimicrobial resistance elements in the gut microbiota.
Potential for intercontinental transfer of antimicrobial drug–resistant bacteria in the microbiota necessitates studies that focus on the antimicrobial resistance of the gut microbiome as a whole, the so-called “gut resistome,” by using culture-independent metagenomic approaches (9). Metagenomic approaches avoid the bias that is introduced when selective culturing is applied because ≈80% of the gut microbiota is not cultivatable (11).
Travel to geographic areas in which rates of bacteria that are resistant to antimicrobial drugs are high has been indicated as a risk factor for the acquisition of such bacteria (12). Studies in Australia (13), Sweden (14,15), and the Netherlands (16) have shown that international travel is a major risk factor for colonization with ESBL-producing Enterobacteriaceae. Likely, these resistant strains are acquired from the environment during travel, e.g., through food consumption (17). Because the human intestinal microbiome will come in contact with many different bacterial species from travel-related environments, the effect of international travel on antimicrobial drug resistance is most likely limited to neither opportunistic pathogens, such as Escherichia coli, nor to ESBL-encoding resistance genes.
In this study, we aimed to investigate the effect of international travel on the human gut resistome. By using a targeted (PCR-based) metagenomic approach, we compared the presence and relative abundance of specific resistance determinants in the entire human gut microbiome before and after international travel.
Population and Design
Healthy long-distance travelers were recruited during November 2010–August 2012 through travel clinics (EASE Travel Clinic & Health Support, www.ease-travelclinic.nl/en/) located in the southern part of the Netherlands. Travelers consenting to participate were asked to collect a fecal sample before and immediately after travel and to provide records of the duration and destination of their travel, illnesses or complaints during travel, drug use, and antimicrobial drug use within the 3 months preceding travel. The fecal samples were sent to clinics by regular mail on the same day of collection and were processed on the day of receipt. The study comprised 122 travelers.
The countries visited were categorized into geographic regions. These regions were Southeast Asia (Asia excluding the Indian subcontinent and the Middle East), the Indian subcontinent (Bangladesh, Bhutan, India, Nepal, Pakistan, and Sri Lanka), northern Africa (countries north of the equator), southern Africa (countries south of the equator), southern Europe, Central America, and South America.
Fecal Specimen Processing and DNA Extraction
Fecal samples were diluted 10-fold in peptone/water solution (Oxoid, Basingstoke, UK) containing 20% (vol/vol) glycerol (Merck, Darmstadt, Germany) and homogenized by vortexing. They were stored at −20°C until molecular analysis was performed.
For the extraction of metagenomic DNA, 200 μL of diluted feces was added to a 2-mL vial containing 0.5 g of 0.1 mm zirconia/silica beads (BioSpec, Bartlesville, OK, USA), 4 glass beads, 3.0–3.5 mm (BioSpec), and 1.2 mL of lysis buffer from the PSP Spin Stool Kit (Stratec Molecular, Berlin, Germany). Samples were disrupted in a Magna Lyser device (Roche, Basel, Switzerland) in 3 cycles of 1 min. at 5,500 rpm. Subsequently, metagenomic DNA was isolated from the samples by using the PSP Spin Stool Kit according to the manufacturer’s instructions. DNA was eluted in 200 μL elution buffer and stored at −20°C until further analysis.
Real-time PCR
Real-time PCR was performed to detect and quantify the β-lactamase–encoding genes cfxA, blaCTX-M, and blaNDM; tetracycline resistance–encoding genes tetM and tetQ; macrolide resistance–encoding gene ermB; aminoglycoside resistance–encoding gene aac(6′)-aph(2′′); and quinolone resistance encoding genes qnrA, qnrB, and qnrS. The 16S rDNA was amplified as a reference gene to normalize for the amount of bacterial DNA in the samples.
The 16S rDNA, cfxA, tetM, tetQ, ermB, and aac(6′)-aph(2′′) targets were amplified by using a MyiQ Single-Color Real-Time PCR Detection System (BioRad, Hercules, CA, USA) in 25-μL reactions containing 12.5 μL iQ SYBR Green Supermix (BioRad) and 5-μL template DNA. Melting curves were checked for each sample to confirm amplification of the correct product. For every target, amplified PCR products of 10 random positive samples were separated by agarose gel electrophoresis to control for purity and size of the amplicons. Finally, for all genes except the 16S rDNA (because of expected heterozygous amplicons), these products were sequenced by using the PCR primers and an ABI BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Sequencing data were obtained by using an ABI 3730 DNA Analyzer (Applied Biosystems) and were analyzed by using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
The blaCTX-M, blaNDM, qnrA, qnrB, and qnrS genes were amplified on a 7900HT Fast Real-Time PCR System (Applied Biosystems) in 25-μL reactions containing 12.5 μL ABsolute QPCR ROX Mix (Thermo Scientific, Waltham, MA, USA) and 10-μL template DNA. The blaCTX-M assay enables identification of the various phylogenetic groups by use of 4 probes. The probes to detect blaCTX-M groups 1 and 2 were combined in the first reaction, and the probe to detect blaCTX-M group 9 was combined with a probe to detect all groups except for the CTX-M-1 group in a second reaction. All primer and probe sequences and PCR conditions for each target are displayed in Table 1.
To determine the efficiency of the PCR, cycle thresholds obtained from a series of 5 template DNA dilutions of at least 3 different samples were graphed on the y-axis versus the log of the dilution on the x-axis. For blaNDM, a clinical isolate was used because no positive fecal samples were available. The PCR efficiencies were 16S rDNA, 94.0%; cfxA, 99.0%; tetM, 97.6%; tetQ, 95.9%; ermB, 95.5%; aac(6’)-aph(2”), 97.0%; blaCTX-M-1+2, 98.2%; blaCTX-M-9+2–8-9–25, 96.7%; blaNDM, 98.4%; qnrA, 97.4%; qnrB, 101.0%; and qnrS, 102.5%.
We determined PCR detection limits for blaCTX-M, qnrB, and qnrS. Clinical isolates harboring these genes were suspended in a 0.5 mol/U McFarland solution, then diluted 10-fold in sterile saline solution. Quantification of CFU in the suspensions was achieved by inoculating blood agar plates (Oxoid) and counting the number of colonies after overnight incubation at 37°C. Next, 20 μL of the quantified suspensions was mixed with 180 μL of feces and submitted to DNA extraction as described above. Subsequently, quantitative PCR was performed on extracted DNA to generate standard curves for quantification. For blaCTX-M, the detection limit was 12–40 CFU/PCR. For qnrB and qnrS, the detection limit was 1–5 CFU/PCR.
Statistical Analyses
We calculated differences in relative resistance gene abundances between samples from before and after travel for each traveler by using the ΔΔCt method with a Pfaffl modification to correct for PCR efficiency (ratio: Etarget^ΔCTtarget/Ereference^ΔCTreference) (24), which is the standard method to measure the relative change in mRNA expression levels by using real-time PCR. However, in this study, rather than measuring mRNA expression levels, the relative amount of target DNA present was measured by using this method. The 16S rDNA was used as the reference gene.
To better visualize increases and decreases in gene abundances in graphs, we converted abundance ratios to a fold change. To determine the overall abundance change of a resistance gene, ratios were log-transformed. A 2-tailed, 1-sample t test was used to test whether the mean log ratio significantly differed from 0.
The number of fecal samples positive for a resistance gene after travel was compared with the positive samples obtained before travel by using the McNemar test for paired samples. Multivariable logistic regression analyses were used to test for the association between age, sex, travel destination and duration, traveler’s diarrhea, and antimicrobial drug use preceding travel (independent variables) and the acquisition of antimicrobial resistance genes (dependent variable). The association between acquisitions of multiple resistance genes was determined by a χ2 test. All analyses were performed by using IBM SPSS Statistics version 20 (www-01.ibm.com/support/docview.wss?uid=swg24029274). Results were interpreted as statistically significant when p was <0.05.
Study Population
The study comprised 122 travelers (71 women, 51 men) whose median age was 43 years (range 18–72 years). The median length of stay abroad was 21 days (range 5–240 days). Fourteen participants traveled for >60 days; 5 participants traveled for >120 days. Most participants visited 1 country; 22 visited >1 country. Six participants visited >1 of the defined geographic regions (Table 2); 7 participants did not provide information about their destination.
Prevalence of Resistance Genes in Fecal Samples
Figure 1
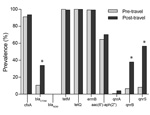
Figure 1. . . Prevalence (%) of antimicrobial drug resistance determinants in fecal samples from 122 healthy travelers from the Netherlands before and after travel, 2010–2012. Statistical significance of the prevalence between the...
Figure 1 shows the prevalence of the antimicrobial drug resistance determinants in fecal samples from the 122 healthy volunteers before and after international travel. The cfxA gene was detected in 111 (91.0%) fecal samples before travel and in 115 (94.3%) samples after travel. The ESBL encoding blaCTX-M gene was prevalent in 11 (9.0%) before travel and in 41 (33.6%) samples after travel, which was a significant increase (p<0.001).
After travel, samples from 5 participants contained blaCTX-M genes of 2 different phylogenetic groups. Before travel, single CTX-M variant was detected for 2 of these persons, and blaCTX-M genes were not detected for the other 3 persons. After travel, the gene was not detected in the samples of 6 persons who were positive for the blaCTX-M gene before travel. The carbapenemase-encoding gene blaNDM was not detected in any sample.
The prevalence of both tetM and tetQ was very high in the fecal samples. The tetM gene was present in all samples before travel and in 121 (99.2%) samples after travel, and tetQ was detected in all samples before and after travel. The prevalence of the ermB gene was also high in samples both before and after travel (99.2% for both). The prevalence of the aac(6′)-aph(2′′); gene was not altered by traveling; this gene was present in 79 (64.5%) of samples before travel and in 86 (70.5%) samples after travel.
Before travel, prevalence of the quinolone resistance genes qnrA, qnrB, and qnrS was relatively low: 0.8%, 6.6%, and 8.2%, respectively. After travel, each of the 3 genes increased: qnrA, qnrB, and qnrS were detected in 3.3%, 36.9%, and 55.7% of samples, respectively. qnrB and qnrS were significantly higher after than before travel (p<0.001).
Relative Gene Abundance Before and After Travel
Figure 2

Figure 2. . . Relative changes in gene abundance before and after travel for each of 122 healthy travelers from the Netherlands during 2010–2012 for genes cfxA (A), tetM (B), tetQ (C), and...
Because the prevalence of the cfxA, tetM, tetQ, and ermB genes was very high before and after travel, we compared the relative abundance of the genes in both samples from each traveler to determine whether traveling influenced the gene abundance. For all 4 genes, the observed changes in gene abundance per traveler were distributed between increases and decreases (Figure 2). Determining the overall increase or decrease of the abundance of each gene showed that none of the investigated genes changed significantly (p>0.05 for all) in abundance after travel.
Effect of Travel Destination and Other Risk Factors on Gene Acquisition
The rate of acquisition of a blaCTX-M gene was highest for travelers visiting the Indian subcontinent (58.1%; p<0.05, OR 26.22, 95% CI 2.86–240.38) (Table 3). Travel to other regions was associated with a blaCTX-M acquisition rate of 17.9% for Southeast Asia and 31.3% and 29.4% for northern and southern Africa, respectively. In the combined category comprising southern Europe, Central America, and South America, 1 blaCTX-M acquisition (6.3%) was detected in a traveler who had been to southern Europe (Turkey).
The acquisition of the qnrB gene was not associated with travel to a specific region, whereas the acquisition of qnrS was highest for Southeast Asia (75.0%; p = 0.001, OR 15.7, 95% CI 3.1–79.2), and second highest for the Indian subcontinent (61.3%; p<0.05, OR 9.2, 95% CI 1.9–43.9). The acquisition rate was also elevated for northern Africa (43.8%) and southern Africa (35.3%) but not significantly so.
We also investigated associations between age, sex, travel destination and duration, traveler’s diarrhea, and antimicrobial drug use preceding the travel and the acquisition of resistance genes. No associations were found (Table 3).
Phylogenetic Groups of blaCTX-M Genes and Association with qnr Genes
Of the 41 blaCTX-M genes acquired during travel, 24 belonged to the CTX-M-1 group, 2 belonged to the CTX-M-2 group, 6 were of the CTX-M-9 group, and 9 were positive for the CTX-M-2–8-9–25 probe but not for the CTX-M-2 or 9 probe, indicating that these genes were in groups 8 or 25. The CTX-M groups acquired per region are shown in Table 4. In contrast, 9/11 CTX-M types detected in the pre-travel samples belonged to the CTX-M-9 group and 2/11 to the CTX-M-1 group.
Associations between the acquisitions of the blaCTX-M and qnr genes were also investigated (Table 5). The acquisition of a blaCTX-M gene was not associated with that of a qnrB (p = 0.305) or qnrS gene (p = 0.080); neither was the gain of a blaCTX-M gene of the CTX-M-1 group, which was the dominant acquired type (58.5%) associated with the acquisition of either qnrB (p = 0.631) or qnrS (p = 0.256).
We used a metagenomic approach to study effects of international travel on part of the resistome of the human gut microbiota. Our results provide insights into the prevalence of the investigated resistance genes in the human gut microbiota and demonstrate high rates of acquisition of the ESBL encoding gene blaCTX-M and quinolone resistance encoding genes qnrB and qnrS related to international travel. The prevalence of these genes increased from 9.0%, 6.6%, and 8.2% before travel to 33.6%, 36.9%, and 55.7% after travel, respectively.
Prospective cohort studies among travelers from Australia (13), the Netherlands (16), and Sweden (14,15) showed that international travel was a risk factor for colonization with ESBL-producing Enterobacteriaceae spp. and that travel to India or the Indian subcontinent was the highest risk factor. These findings agree with the rates of blaCTX-M acquisition found in our study, which were highest for travelers to the Indian subcontinent.
The phylogenetic types of the blaCTX-M gene that were acquired in our study group were clearly dominated by CTX-M group 1, especially in the Indian subcontinent. This geographical association corresponds to the aforementioned cohort studies (13–16), which showed that ESBL-producing Enterobacteriaceae identified in travelers to India or the Indian subcontinent mainly comprise CTX-M group 1. Although the statistical power of our study was insufficient to analyze the specific CTX-M groups, it was striking that genes of the CTX-M-2 group were detected twice and those of either group 8 or 25 were detected 9 times. In previous studies, these CTX-M groups were not detected at all (13,14) or were detected only sporadically (15,16). The difference in results could be caused by our use of a metagenomic approach, which might detect blaCTX-M in a much wider array of species than did studies investigating specific cultured Enterobacteriaceae spp. This difference in approach might furthermore explain that of the blaCTX-M genes detected before travel in the population in our study, most (9/11, 82%) were of the CTX-M-9 group, which contrasts studies that report that blaCTX-M-15 (which belongs to the CTX-M-1 group) is predominant in ESBL-producing Enterobacteriaceae in the Netherlands (16,25,26). Aside from the different method used, the population sizes in these studies were larger than the cohort in our current study.
Plasmid-mediated quinolone resistance genes, such as the qnr variants, provide low-level quinolone resistance. However, these genes are relevant because they facilitate the emergence of higher-level resistance and thus can speed the development and spread of resistance to these antimicrobial agents (27). Although foreign travel has been associated with the acquisition of plasmid-mediated quinolone resistant–positive isolates (28–30), these genes have thus far not been focused on in prospective cohort studies investigating the effects of travel on antimicrobial resistance.
A study by Vien et al. that investigated the prevalence of the qnr genes in fecal swab samples from children in Vietnam who had acute respiratory tract infections (23) showed very high qnrS prevalence (74.5%). Travel to areas with such a high prevalence could be a major risk factor for acquisition of these genes. Five (83%) of 6 participants in our study who had traveled to Vietnam acquired a qnrS gene. In total, 11 volunteers had traveled to Cambodia, Thailand, Vietnam, or a combination of these geographically neighboring countries, and 9 (82%) acquired a qnrS gene. These data suggest that organisms carrying the qnrS gene are highly prevalent in these areas and that travelers visiting these areas have a high risk for exposure to those organisms.
Coexistence of qnr genes with various other resistance genes, such as blaCTX-M, on the same plasmid is well known (31–34) and could be related to our finding that both types of genes were more prevalent in the study participants’ samples after travel. However, we found no association between these genes in these samples. The qnrS gene was most often acquired by travelers who visited Southeast Asia and, to a lesser extent, the Indian subcontinent, whereas the acquisition rate for blaCTX-M was clearly highest for travelers to the Indian subcontinent but was not higher for travelers to Southeast Asia than for travelers to other regions. These findings indicate that although travel to the Indian subcontinent is a high-risk factor for acquiring both of these genes, these risk factors are not necessarily related.
Compared with culturing methods, a metagenomic approach has the advantage of being able to detect resistance in a much wider array of species; however, a limitation is that it is not yet known in which organisms the acquired resistance genes detected in our study are present, nor if they are being expressed. Another limitation of our study is that the study population was not large enough for us to conduct a more extensive risk analysis. Future studies that conduct more extensive analyses for risk factors, such as antimicrobial drug use, travel destination, and duration of travel, would benefit from larger populations. Furthermore, in future studies inclusion of a follow-up sampling of travelers would be highly relevant for investigating the period in which these acquired resistance genes remain in the resistome and if the perseverance or even HGT of these genes in the resistome is promoted by factors such as selective pressure introduced by antimicrobial drug use. Little is known about the duration of travel-acquired resistant organisms in the human microbiota, although their continued viability plays a key role in the ability to further spread these organisms or resistance elements.
During our investigation of several targeted resistance genes, it became evident that resistance genes from foreign environments are being introduced into the gut resistome at high rates related to international travel. Although the consequences of these changes in the resistome are difficult to predict, the introduction of these genes into the genetic pool of resistance elements may create opportunities for the horizontal transfer to other organisms in the gut microbiota.
Our study data demonstrated an increasing prevalence of blaCTX-M, qnrB, and qnrS genes in the feces of healthy volunteers from the Netherlands immediately after they returned from international travel. These findings contribute to the increasing evidence that travelers contribute to the spread of antimicrobial drug resistance.
Mr von Wintersdorff is a PhD student at the Department of Medical Microbiology at the Maastricht University Medical Center, the Netherlands. His research interests include the microbial resistome and the horizontal gene transfer of antimicrobial resistance.
Acknowledgment
We thank Mayk Lucchesi for his work in the processing and storage of the fecal samples.
References
- Wright GD. The antibiotic resistome. Expert Opinion on Drug Discovery. 2010;5:779–88.PubMedGoogle Scholar
- Zhou W, Wang Y, Lin J. Functional cloning and characterization of antibiotic resistance genes from the chicken gut microbiome. Appl Environ Microbiol. 2012;78:3028–32. DOIPubMedGoogle Scholar
- Cheng G, Hu Y, Yin Y, Yang X, Xiang C, Wang B, Functional screening of antibiotic resistance genes from human gut microbiota reveals a novel gene fusion. FEMS Microbiol Lett. 2012;336:11–6. DOIPubMedGoogle Scholar
- Torres-Cortés G, Millan V, Ramirez-Saad HC, Nisa-Martinez R, Toro N, Martinez-Abarca F. Characterization of novel antibiotic resistance genes identified by functional metagenomics on soil samples. Environ Microbiol. 2011;13:1101–14. DOIPubMedGoogle Scholar
- Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science. 2009;325:1128–31. DOIPubMedGoogle Scholar
- Wright GD. Antibiotic resistance in the environment: a link to the clinic? Curr Opin Microbiol. 2010;13:589–94. DOIPubMedGoogle Scholar
- Poirel L, Rodriguez-Martinez JM, Mammeri H, Liard A, Nordmann P. Origin of plasmid-mediated quinolone resistance determinant qnrA. Antimicrob Agents Chemother. 2005;49:3523–5. DOIPubMedGoogle Scholar
- Poirel L, Kampfer P, Nordmann P. Chromosome-encoded ambler class a beta-lactamase of Kluyvera georgiana, a probable progenitor of a subgroup of CTX-M extended-spectrum beta-lactamases. Antimicrob Agents Chemother. 2002;46:4038–40. DOIPubMedGoogle Scholar
- Penders J, Stobberingh EE, Savelkoul PH, Wolffs PF. The human microbiome as a reservoir of antimicrobial resistance. Frontiers in Microbiology. 2013;4:87.
- Baquero F. Metagenomic epidemiology: a public health need for the control of antimicrobial resistance. Clin Microbiol Infect. 2012;18(Suppl 4):67–73. DOIPubMedGoogle Scholar
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Diversity of the human intestinal microbial flora. Science. 2005;308:1635–8. DOIPubMedGoogle Scholar
- van der Bij AK, Pitout JD. The role of international travel in the worldwide spread of multiresistant enterobacteriaceae. J Antimicrob Chemother. 2012;67:2090–100. DOIPubMedGoogle Scholar
- Kennedy K, Collignon P. Colonisation with Escherichia coli resistant to “critically important” antibiotics: a high risk for international travellers. Eur J Clin Microbiol Infect Dis. 2010;29:1501–6. DOIPubMedGoogle Scholar
- Tängdén T, Cars O, Melhus A, Lowdin E. Foreign travel is a major risk factor for colonization with Escherichia coli producing CTX-M–type extended-spectrum beta-lactamases: a prospective study with Swedish volunteers. Antimicrob Agents Chemother. 2010;54:3564–8. DOIPubMedGoogle Scholar
- Ostholm-Balkhed A, Tarnberg M, Nilsson M, Nilsson LE, Hanberger H, Hallgren A, Travel-associated faecal colonization with ESBL-producing enterobacteriaceae: Incidence and risk factors. J Antimicrob Chemother. 2013;68:2144–53 . DOIPubMedGoogle Scholar
- Paltansing S, Vlot JA, Kraakman MEM, Mesman R, Bruijning ML, Bernards AT, Extended-spectrum β-lactamase–producing enterobacteriaceae among travelers from the Netherlands. Emerg Infect Dis. 2013;19:1206–13. DOIPubMedGoogle Scholar
- Collignon P. Resistant Escherichia coli—we are what we eat. Clin Infect Dis. 2009;49:202–4 . DOIPubMedGoogle Scholar
- Vliegen I, Jacobs JA, Beuken E, Bruggeman CA, Vink C. Rapid identification of bacteria by real-time amplification and sequencing of the 16s rRNA gene. J Microbiol Methods. 2006;66:156–64. DOIPubMedGoogle Scholar
- Kim SM, Kim HC, Lee SW. Characterization of antibiotic resistance determinants in oral biofilms. J Microbiol. 2011;49:595–602. DOIPubMedGoogle Scholar
- Martineau F, Picard FJ, Lansac N, Menard C, Roy PH, Ouellette M, Correlation between the resistance genotype determined by multiplex PCR assays and the antibiotic susceptibility patterns of Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob Agents Chemother. 2000;44:231–8. DOIPubMedGoogle Scholar
- Birkett CI, Ludlam HA, Woodford N, Brown DF, Brown NM, Roberts MT, Real-time TaqMan PCR for rapid detection and typing of genes encoding CTX-M extended-spectrum beta-lactamases. J Med Microbiol. 2007;56:52–5. DOIPubMedGoogle Scholar
- Naas T, Ergani A, Carrer A, Nordmann P. Real-time PCR for detection of NDM-1 carbapenemase genes from spiked stool samples. Antimicrob Agents Chemother. 2011;55:4038–43. DOIPubMedGoogle Scholar
- Vien LTM, Minh NN, Thuong TC, Khuong HD, Nga TV, Thompson C, The co-selection of fluoroquinolone resistance genes in the gut flora of Vietnamese children. PLoS ONE. 2012;7:e42919. DOIPubMedGoogle Scholar
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. DOIPubMedGoogle Scholar
- Reuland EA, Overdevest IT, Al Naiemi N, Kalpoe JS, Rijnsburger MC, Raadsen SA, High prevalence of ESBL-producing enterobacteriaceae carriage in Dutch community patients with gastrointestinal complaints. Clin Microbiol Infect. 2013;19:542–9. DOIPubMedGoogle Scholar
- Leverstein-van Hall MA, Dierikx CM, Cohen Stuart J, Voets GM, van den Munckhof MP, van Essen-Zandbergen A, Dutch patients, retail chicken meat and poultry share the same ESBL genes, plasmids and strains. Clin Microbiol Infect. 2011;17:873–80. DOIPubMedGoogle Scholar
- Martínez-Martínez L, Pascual A, Jacoby GA. Quinolone resistance from a transferable plasmid. Lancet. 1998;351:797–9. DOIPubMedGoogle Scholar
- Hopkins KL, Wootton L, Day MR, Threlfall EJ. Plasmid-mediated quinolone resistance determinant qnrs1 found in Salmonella enterica strains isolated in the UK. J Antimicrob Chemother. 2007;59:1071–5. DOIPubMedGoogle Scholar
- Karczmarczyk M, Stephan R, Hachler H, Fanning S. Complete nucleotide sequence of pVQS1 containing a quinolone resistance determinant from Salmonella enterica serovar virchow associated with foreign travel. J Antimicrob Chemother. 2012;67:1861–4. DOIPubMedGoogle Scholar
- Taguchi M, Kawahara R, Seto K, Inoue K, Hayashi A, Yamagata N, Plasmid-mediated quinolone resistance in Salmonella isolated from patients with overseas travelers' diarrhea in Japan. Jpn J Infect Dis. 2009;62:312–4 http://www0.nih.go.jp/JJID/62/312.html.PubMedGoogle Scholar
- Kanamori H, Yano H, Hirakata Y, Hirotani A, Arai K, Endo S, Molecular characteristics of extended-spectrum beta-lactamases and qnr determinants in enterobacter species from Japan. PLoS ONE. 2012;7:e37967. DOIPubMedGoogle Scholar
- Strahilevitz J, Jacoby GA, Hooper DC, Robicsek A. Plasmid-mediated quinolone resistance: a multifaceted threat. Clin Microbiol Rev. 2009;22:664–89. DOIPubMedGoogle Scholar
- Jiang Y, Zhou Z, Qian Y, Wei Z, Yu Y, Hu S, Plasmid-mediated quinolone resistance determinants qnr and aac(6')-ib-cr in extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae in China. J Antimicrob Chemother. 2008;61:1003–6. DOIPubMedGoogle Scholar
- Dahmen S, Poirel L, Mansour W, Bouallegue O, Nordmann P. Prevalence of plasmid-mediated quinolone resistance determinants in enterobacteriaceae from Tunisia. Clin Microbiol Infect. 2010;16:1019–23.PubMedGoogle Scholar
Figures
Tables
Cite This Article*These authors contributed equally to this article and are co–first authors.
Table of Contents – Volume 20, Number 4—April 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Petra F.G. Wolffs, Department of Medical Microbiology, Maastricht University Medical Center, PO Box 5800, 6202 AZ, Maastricht, the Netherlands
Top