Volume 22, Number 4—April 2016
Dispatch
Hypervirulent emm59 Clone in Invasive Group A Streptococcus Outbreak, Southwestern United States
Cite This Article
Citation for Media
Abstract
The hyper-virulent emm59 genotype of invasive group A Streptococcus was identified in northern Arizona in 2015. Eighteen isolates belonging to a genomic cluster grouped most closely with recently identified isolates in New Mexico. The continued transmission of emm59 in the southwestern United States poses a public health concern.
Several cases of invasive group A Streptococcus (GAS) disease were detected in January 2015 in a northern Arizona hospital. A substantive percentage of the cases were associated with a homeless shelter and a local jail; outbreak case-patients were predominantly male and Native American. Other studies have shown an increase in infection risk for invasive GAS in Native American/First Nations populations (1,2), and outbreaks within this population in Arizona have been previously documented (3). Whole genome sequence analysis determined that the hypervirulent subtype emm59 was present among the first cases analyzed in early 2015. emm59 is known to have caused a nationwide outbreak of invasive GAS in Canada during 2006–2009 (4,5), and cases and outbreaks have been reported in the United States (6–8).
We identified isolates for sequencing from 29 invasive GAS cases diagnosed in patients in a northern Arizona hospital during January–July 2015 and randomly selected an additional 99 GAS isolates from a repository of >2,000 Arizona GAS isolates collected during 2002–2006 (no isolates from patients in Arizona were available for 2007–2014). Four additional isolates from central Arizona identified in 2015 were included in the analysis (Technical Appendix Table). All isolates were grown on 5% sheep blood tryptic soy agar plates (Hardy Diagnostics, Santa Maria, CA), and incubated at 37°C with 5% CO2. DNA was extracted by using a DNeasy Blood and Tissue Kit (QIAGEN, Valencia, CA, USA) following manufacturer’s protocol. Genomic DNA libraries were prepared by using the Nextera XT library prep kit (Illumina, San Diego, CA) and sequenced with paired-end reads (250 bp) on an Illumina MiSeq instrument, as previously described (9). The finished genome of the emm59 Canadian clone MGAS15252 (GenBank accession no. CP003116) and high-quality publicly available sequence-read data from 44 US isolates, from NCBI short read archive (BioProject #PRJNA194066), were included in the subsequent phylogenetic analyses. The final core genome (all nucleotide loci found in all genomes) for single-nucleotide polymorphism (SNP) detection was 1,636,024 bp (98.6% of reference).
We used NASP SNP analysis pipeline (http://tgennorth.github.io/NASP/) for whole-genome SNP typing as previously described (10). SNP matrices were developed for both the whole species and the emm59-only analyses. We used MEGA version 5.2.2 software (11) to generate maximum parsimony phylogenetic trees. Regions of high SNP density were identified as possible regions of recombination and were further analyzed for impact on the consistency index. Genomes were assembled by using UGAP (https://github.com/jasonsahl/UGAP). GAS emm subtypes were assigned by using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi), querying the study genome assemblies against the Centers for Disease Control and Prevention’s (CDC) emm type-specific sequence database (http://www.cdc.gov/streplab/m-proteingene-typing.html). We resolved dual emm-type hits using CDC’s emm typing Sanger sequencing primers (http://www.cdc.gov/streplab/protocol-emm-type.html) as a BLAST query and noting hit locations.
We identified 18 of the 29 contemporary northern Arizona isolates as subtype emm59; the remaining isolates were composed of 6 additional emm types: emm1 (n = 2), emm5 (n = 2), emm58 (n = 1), emm81 (n = 2), emm83 (n = 1), emm89 (n = 2), and emm94 (n = 1). The 99 historical and 4 contemporary background Arizona isolates included 25 distinct emm types (Technical Appendix Table). No emm59 isolates were identified in this background set, and none had been previously reported in Arizona.
Figure 1

Figure 1. Phylogenetic single-nucleotide polymorphism (SNP) tree of emm59 isolates from a northern Arizona hospital displaying distribution of mutations in a 23kb positively selected region during invasive group A Streptococcus outbreak, southwestern United...
The 18 Arizona emm59 cases occurred during January–July 2015 (Table). An emm59-only phylogenetic analysis demonstrated the apparent presence of multiple lineages of emm59 in the 2015 Arizona isolates (Figure 1). A distinct clone consisting of 14 of the 18 emm59 isolates were separated from each other by only 0–4 SNPs, genomically supporting the presence of an ongoing outbreak; >8 of these patients were epidemiologically linked to physical contact, cohabitation, or both with 1 other person (data not shown). The additional emm59 isolates make up additional lineages separated from one other by 8–28 SNPs. No recombination was identified among the Arizona isolates. A relatively large number of SNPs and indels were seen within an approximate 23-kilobase region (Figure 1). This region has been previously reported to contain mutational hotspots associated with virulence (12,13). Considering the presumptive positive selective force on this region, SNPs within the region were not included in the final phylogenetic analysis.
Figure 2
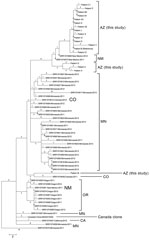
Figure 2. Phylogenetic single-nucleotide polymorphism (SNP) tree of emm59 isolates from Arizona during invasive group A Streptococcus outbreak in the southwestern United States, previously analyzed US emm59 isolates, and the Canadian clone. Maximum...
When compared with all other publicly available US emm59 isolate genomes, nearly all the genomes identified in the United States were closely related to each other and to the Canadian clone MGAS15252; individual isolate SNP branch lengths ranged from 0 to 10 (Figure 2). The Arizona outbreak isolates were separated from 2 New Mexico isolates by 4 and 5 SNPs each; these isolates fell within the overall Arizona clade and were subsequently included in the Arizona-only phylogenetic analysis (Figure 1). Conversley, the isolate from patient M appears more distant from the larger Arizona population. The Arizona clades, with the exception of that of the isolate from patient M, all appear to arise from the large Minnesota polytomy. The previously estimated 1.3–2.1 SNPs/year mutation rates for GAS (14,15) further support the Arizona outbreak as being caused by a single clone, likely originating from New Mexico and being spread over 6–12 months.
The emm59 subtype of GAS, the etiologic agent of a substantial nationwide outbreak of invasive GAS in Canada during 2006–2009 (4), is now present in Arizona, causing at least 1 outbreak of epidemiologically and genomically linked cases and several additional epidemiologically unrelated cases. The lack of emm59 in background isolates in Arizona from the previous decade, along with its low genetic diversity, suggests that emm59 emerged recently in Arizona. Following the emm59 epidemic in Canada, this subtype was subsequently seen in a few US states; a retrospective analyses of the Centers for Disease Control and Prevention Active Bacterial Core surveillance (ABCs) system (http://www.cdc.gov/abcs/reports-findings/survreports.pdf) identified 40 US emm59 isolates during 2000–2009 (6) and an additional 67 isolates during 2010–2012 (7). Of note, only 5 (of the 40 emm59 isolates from 2000–2009 (2 from Minnesota, 2 from California, and 1 from Oregon) appeared to be closely related to the Canadian clone (defined by the authors as being separated by <16 SNPs) (6); in contrast, all of the strains from the 2010–2012 survey appeared to be more closely related to the Canadian clone. The more recent ABCs analysis identified an increasing number of southwestern isolates, including 4 from Colorado and 6 from New Mexico (7), although no outbreaks were specifically described in these states (Arizona is not included in the ABCs system). Similar to this outbreak study, Olsen et al. (7), in an analysis of 60 MN emm59 isolates from case-patients with identified race, determined that 25 (42%) were from Native Americans; of 5 isolates from New Mexico in that study, 3 were from Native Americans.
Given the apparent distal nature of the Arizona/New Mexico isolates to the Minnesota population in our study, it is reasonable to propose an unidentified epidemiologic relationship between these case populations. However, caution must be used in drawing conclusions regarding the relationships of isolates from disparate geographic regions because only limited comparable sequence data from previous emm59 studies in the United States (7) were publicly available to compare to the Arizona isolates. Epidemiologic investigations, along with healthcare provider and patient education activities, are ongoing in Arizona to further determine the extent of the current outbreak and the associated risk factors and to help mitigate effects and limit or prevent further spread to at-risk populations.
Dr. Engelthaler is an associate professor with the Translational Genomics Research Institute in Flagstaff, AZ. His research interests are in advancing epidemiology and clinical response through applied infectious disease genomics.
References
- Hoge CW, Schwartz B, Talkington DF, Breiman RF, MacNeill EM, Englender SJ. The changing epidemiology of invasive group A streptococcal infections and the emergence of streptococcal toxic shock–like syndrome. A retrospective population-based study. JAMA. 1993;269:384–9. DOIPubMedGoogle Scholar
- Athey TB, Teatero S, Sieswerda LE, Gubbay JB, Marchand-Austin A, Li A, High incidence of invasive group A Streptococcus disease caused by strains of uncommon emm types in Thunder Bay, Ontario, Canada. J Clin Microbiol. 2016;54:83–92 .DOIPubMedGoogle Scholar
- Harris AM, Yazzie D, Antone-Nez R, Dinè-Chacon G, Kinlacheeny JB, Foley D, Community-acquired invasive GAS disease among Native Americans, Arizona, USA, Winter 2013. Emerg Infect Dis. 2015;21:177–9. DOIPubMedGoogle Scholar
- Fittipaldi N, Beres SB, Olsen RJ, Kapur V, Shea PR, Watkins ME, Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A Streptococcus. Am J Pathol. 2012;180:1522–34. DOIPubMedGoogle Scholar
- Tyrrell GJ, Lovgren M, Ibrahim Q, Garg S, Chui L, Boone TJ, Epidemic of invasive pneumococcal disease, western Canada, 2005–2009. Emerg Infect Dis. 2012;18:733–40. DOIPubMedGoogle Scholar
- Fittipaldi N, Olsen RJ, Beres SB, Van Beneden C, Musser JM. Genomic analysis of emm59 group A Streptococcus invasive strains, United States. Emerg Infect Dis. 2012;18:650–2. DOIPubMedGoogle Scholar
- Olsen RJ, Fittipaldi N, Kachroo P, Sanson MA, Long SW, Como-Sabetti KJ, Clinical laboratory response to a mock outbreak of invasive bacterial infections: a preparedness study. J Clin Microbiol. 2014;52:4210–6. DOIPubMedGoogle Scholar
- Brown CC, Olsen RJ, Fittipaldi N, Mormon ML, Fort PL, Neuwirth R, Spread of virulent group A Streptococcus type emm59 from Montana to Wyoming, USA. Emerg Infect Dis. 2014;20:679–81. DOIPubMedGoogle Scholar
- Driebe EM, Sahl JW, Roe CC, Bowers JR, Schupp JM, Gillece JD, Using whole genome analysis to examine Recombination Across Diverse Sequence Types of Staphylococcus aureus. PLoS ONE. 2015;10:e0130955. DOIPubMedGoogle Scholar
- Struve C, Roe CC, Stegger M, Stahlhut SG, Hansen DS, Engelthaler DM, Mapping the evolution of hypervirulent Klebsiella pneumoniae. MBio. 2015;6:e00630 . DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using Streptococcus, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. DOIPubMedGoogle Scholar
- Fittipaldi N, Tyrrell GJ, Low DE, Martin I, Lin D, Hari KL, Integrated whole-genome sequencing and temporospatial analysis of a continuing group A Streptococcus epidemic. Emerg Microbes Infect. 2013;2:e13.
- Sanson M, O’Neill BE, Kachroo P, Anderson JR, Flores AR, Valson C, A naturally occurring single amino acid replacement in multiple gene regulator of group A Streptococcus significantly increases virulence. Am J Pathol. 2015;185:462–71 . DOIPubMedGoogle Scholar
- Nasser W, Beres SB, Olsen RJ, Dean MA, Rice KA, Long SW, Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc Natl Acad Sci U S A. 2014;111:E1768–76. DOIPubMedGoogle Scholar
- Turner CE, Abbott J, Lamagnic T, Holden MTG, David S, Jones MD, Emergence of a new highly successful acapsular group A Streptococcus clade of the genotype emm89. MBio. 2015;6:e00622–15 .DOIPubMedGoogle Scholar
Figures
Table
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 22, Number 4—April 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
David M. Engelthaler, TGen North, The Translational Genomics Research Institute, 3051 W. Shamrell Blvd., Suite 106, Flagstaff, AZ 86001, USA
Top