Volume 18, Number 12—December 2012
Dispatch
Differentiation of Prions from L-type BSE versus Sporadic Creutzfeldt-Jakob Disease
Cite This Article
Citation for Media
Abstract
We compared transmission characteristics for prions from L-type bovine spongiform encephalopathy and MM2-cortical sporadic Creutzfeldt-Jakob disease in the Syrian golden hamster and an ovine prion protein–transgenic mouse line and isolated distinct prion strains. Our findings suggest the absence of a causal relationship between these diseases, but further investigation is warranted.
Among transmissible spongiform encephalopathies (TSEs), the L-type bovine spongiform encephalopathy (L-BSE) in cattle requires particular attention for public health. L-BSE is transmitted more efficiently than is classical BSE among primates (1–3) as well as among transgenic mice that express human prion protein (PrP) (4,5). We recently reported that L-BSE was readily transmissible by experimental oral inoculation in a nonhuman primate species, the grey mouse lemur (Microcebus murinus) (3). These findings raise the possibility that some human Creutzfeldt-Jakob disease (CJD) cases might result from exposure to the L-BSE agent; previous studies highlighted similarities between L-BSE and some human subtypes (type 2) of sporadic CJD (sCJD) (1,6).
To examine the possible relationship between L-BSE and sCJD, we evaluated a strain-typing strategy that relies on comparative transmission characteristics in the Syrian golden hamster and in a transgenic mouse line (TgOvPrP4) expressing ovine PrP (ARQ allele). Both of these species are susceptible to L-BSE prions from cattle (7,8). The transmission of L-BSE, including after a first passage in Microcebus murinus lemurs (3), was compared with that for the MM2-cortical subtype of sCJD (9); this subtype was chosen on the basis of a study that indicated higher levels of molecular similarities of L-BSE with this sCJD subtype than with the MV2 subtype (1).
The TSE brain inocula used in this study, conducted during November 2010–December 2011, were derived from 2 natural L-BSE isolates from France (02-2528 and 08-0074); a lemur injected intracerebrally (i.c.) with the 02-2528 L-BSE cattle isolate (3); and a human patient with MM2-cortical sCJD. Consent was obtained for using tissues from the human patient in research, including genetic analyses. Animal experiments were performed in the biohazard prevention area (A3) of the Anses-Lyon animal facilities, in accordance with the guidelines of the French Ethical Committee (decree 87-848) and European Community Directive 86/609/EEC.
Six-week-old TgOvPrP4 mice and 4-week-old Syrian golden hamsters were injected i.c. with 20 and 30 µL, respectively, of 10% (wt/vol) brain homogenates in 5% sterile glucose. Serial passages were performed in TgOvPrP4 mice by i.c. inoculation of 1% (wt/vol) homogenates from mice positive for protease-resistant PrP (PrPres). At the terminal stage of the disease, animals were euthanized, and their brains and spleens were collected for PrPres analyses by Western blot and for histopathologic studies (8).
Figure 1
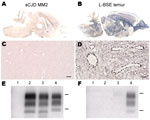
Figure 1. . . . Susceptibility of Syrian golden hamsters to MM2-cortical subtype sporadic Creutzfeldt-Jakob disease (sCJD) and L-type bovine spongiform encephalopathy (L-BSE) prions. Disease-associated prion protein (PrPd) was analyzed in brains of...
In hamsters, transmission of the MM2-cortical sCJD agent was inefficient. Clinical signs were absent up to 876 days postinoculation (dpi) (Table), and disease-associated PrP (PrPd) in brain samples was not detected by paraffin-embedded tissue blot (PET-blot) (Figure 1, panel A), immunohistochemical (Figure 1, panel C), or Western blot (Figure 1, panels E, F) analyses. PrPres was also undetectable in spleen tissues by Western blot (Table).
In contrast, the L-BSE agent passaged in a lemur was efficiently transmitted to hamsters, with a mean survival period of 529 ± 117 dpi, similar to that for L-BSE from cattle (622 ± 64 dpi) (Table). PET-blot analysis (Figure 1, panel B) showed widespread PrPres distribution in the brain; immunohistochemical analysis (Figure 1, panel D) showed a granular type of PrPd deposition that redefined the periphery of most of the blood vessels. Western blot analysis (Figure 1, panels E, F) showed PrPres in the brains of hamsters inoculated with L-BSE from cattle and lemur and in 1/4 spleens of hamsters injected with L-BSE passaged in lemur (Table). Brain PrPres was characterized by low apparent molecular mass (≈19 kDa for the unglycosylated band) associated with a lack of reactivity toward the N terminal 12B2 antibody, in contrast to that for the control animal with scrapie (Figure 1, panels E, F).
In TgOvPrP4 mice, all TSEs were efficiently transmitted, as confirmed by PrPd accumulation in the mouse brains (Table). After serial passages in additional TgOvPrP4 mice, the survival periods in each experiment became considerably shorter (Table; Technical Appendix Figure 1). No statistically significant differences in results were identified between the L-BSE sources (p>0.6). Mean survival period decreased to 111 ± 25 dpi at second passage in mice inoculated with the agent of MM2-cortical subtype sCJD, which differed significantly from that of mice inoculated with L-BSE (p<0.0001). A third passage of both cattle L-BSE and human sCJD did not reduce the survival periods in TgOvPrP4 mice (data not shown).
Figure 2

Figure 2. . . . Western blot molecular typing of protease-resistant prion protein (PrPres) in brain and spleen tissues of ovine prion protein–transgenic (TgOvPrP4) mice at second passage. PrPres from mice infected with...
Western blot analyses of PrPres from mouse brains showed partially similar features for MM2-cortical sCJD and L-BSE, including low molecular mass (≈19 kDa for the unglycosylated band) (Figure 2, panel A) and similar conformational stability of PrPd after treatment with guanidinium hydrochloride (Technical Appendix Figure 2). However, the proportions of diglycosylated, monoglycosylated, and unglycosylated bands of brain PrPres differed between sCJD and L-BSE (Figure 2, panel C); higher proportions of diglycosylated PrPres were found in sCJD-infected mice (mean 67% of the total signal) compared with L-BSE–infected mice (≈18% lower; p<0.0001). PrPres was readily identified in the spleens of TgOvPrP4 mice at the second passage for sCJD and L-BSE from cattle and at the first passage for L-BSE from lemur (Table). No significant differences in the proportions of PrPres glycoforms for sCJD-infected versus L-BSE–infected mice were observed in the spleens (Figure 2, panel D), but PrPres was ≈0.5 kD higher in mice injected with sCJD (Figure 2, panel B, arrows).
Histopathologic analysis showed severe vacuolar lesions in TgOvPrP4 mice infected at second passage with sCJD and lemur-passaged L-BSE (Technical Appendix Figure 3). However, in sCJD-infected mice, vacuolar lesions were mostly observed in the anterior parts of the brain (except the parietal cortex), whereas in mice infected with lemur-passaged L-BSE, the lesions were more widely distributed, involving the colliculi and the hypothalamus. In mice infected with sCJD and lemur-passaged L-BSE, PET-blot analyses showed that most of the PrPres occurred in the frontal parts of the brain, but the intensity and appearance of PrPres in the cortex, thalamus, and hippocampus were distinctly different. Immunohistochemical analyses of the hippocampus showed PrPd deposition in the dentate gyrus in sCJD-infected mice, in contrast to a lack of deposition in lemur-passaged L-BSE–infected mice.
We report the isolation of 2 prion strains derived from L-BSE and MM2-cortical sCJD after transmission in Syrian hamsters and ovine PrP–transgenic mice. In hamsters, we did not transmit any disease with sCJD, but the transmission of L-BSE from lemur was efficient, as previously reported for L-BSE from cattle (7,11). This result suggests that L-BSE did not undergo major modifications after this cross-species transmission and could indicate a clear biologic difference between MM2-cortical sCJD and L-BSE. We also demonstrated the efficient transmission of both L-BSE and MM2-cortical sCJD in TgOvPrP4 mice, which enabled us to compare these diseases in a single model. Unexpectedly, during serial passages, we observed that the agent of MM2-cortical sCJD causes a much more rapidly fatal disease. Despite similar molecular features in sCJD and L-BSE, including the PrPres electrophoretic mobility and the conformational stability of PrPd, sCJD and L-BSE differed in PrPres glycosylation for the mouse brains and gel migrations for the mouse spleens. Mice infected with MM2-cortical sCJD versus those infected with L-BSE also showed distinct lesion profiles and PrPd distribution, which confirms clear biologic differences between these diseases.
Although only 1 case of sCJD of a unique molecular subtype was examined in our study, our observations do not support the hypothesis of a causal relationship between L-BSE and this human sCJD subtype. Our study thus encourages further investigations using the proposed bioassay approach for a more complete evaluation of possible relationships between L-BSE and human prion diseases.
Mr Nicot is a PhD student at the Agence Nationale de Sécurité Sanitaire in Lyon. His primary research interests include characterization of the infectious agents and prion protein during intra- and interspecies transmission of animal and human prion diseases.
Acknowledgments
We thank the staff of the Plateforme d’Expérimentation Animale of Anses-Lyon for excellent animal care; Mikaël Leboidre and Jean-Michel Bridon for histotechnical assistance; Dominique Canal and Claire Aufauvre for biochemical assistance; and Françoise Didier and Nathalie Streichenberger for characterization of the human sporadic CJD case.
S.N. was supported by grants from the Agence Nationale de Sécurité Sanitaire and the Fondation pour la Recherche Médicale.
References
- Comoy EE, Casalone C, Lescoutra-Etchegaray N, Zanusso G, Freire S, Marce D, Atypical BSE (BASE) transmitted from asymptomatic aging cattle to a primate. PLoS ONE. 2008;3:e3017. DOIPubMedGoogle Scholar
- Ono F, Tase N, Kurosawa A, Hiyaoka A, Ohyama A, Tezuka Y, Atypical L-type bovine spongiform encephalopathy (L-BSE) transmission to cynomolgus macaques, a non-human primate. Jpn J Infect Dis. 2011;64:81–4 .PubMedGoogle Scholar
- Mestre-Francés N, Nicot S, Rouland S, Biacabe AG, Quadrio I, Perret-Liaudet A, Oral transmission of L-type bovine spongiform encephalopathy in primate model. Emerg Infect Dis. 2012;18:142–5. DOIPubMedGoogle Scholar
- Kong Q, Zheng M, Casalone C, Qing L, Huang S, Chakraborty B, Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol. 2008;82:3697–701. DOIPubMedGoogle Scholar
- Béringue V, Herzog L, Reine F, Le Dur A, Casalone C, Vilotte JL, Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis. 2008;14:1898–901. DOIPubMedGoogle Scholar
- Casalone C, Zanusso G, Acutis P, Ferrari S, Capucci L, Tagliavini F, Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A. 2004;101:3065–70. DOIPubMedGoogle Scholar
- Nicot S, Baron T. Strain-specific barriers against bovine prions in hamsters. J Virol. 2011;85:1906–8. DOIPubMedGoogle Scholar
- Baron T, Bencsik A, Biacabe AG, Morignat E, Bessen RA. Phenotypic similarity of transmissible mink encephalopathy in cattle and L-type bovine spongiform encephalopathy in a mouse model. Emerg Infect Dis. 2007;13:1887–94. DOIPubMedGoogle Scholar
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33. DOIPubMedGoogle Scholar
- Baron T, Bencsik A, Morignat E. Prions of ruminants show distinct splenotropisms in an ovine transgenic mouse model. PLoS ONE. 2010;5:e10310. DOIPubMedGoogle Scholar
- Shu Y, Masujin K, Okada H, Iwamaru Y, Imamura M, Matsuura Y, Characterization of Syrian hamster adapted prions derived from L-type and C-type bovine spongiform encephalopathies. Prion. 2011;5:103–8. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 18, Number 12—December 2012
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Thierry Baron, Anses-Lyon, 31 Avenue Tony Garnier, 69364 Lyon Cedex 07, France
Top