Volume 19, Number 5—May 2013
Research
Foodborne Transmission of Bovine Spongiform Encephalopathy to Nonhuman Primates
Cite This Article
Citation for Media
Abstract
Risk for human exposure to bovine spongiform encephalopathy (BSE)–inducing agent was estimated in a nonhuman primate model. To determine attack rates, incubation times, and molecular signatures, we orally exposed 18 macaques to 1 high dose of brain material from cattle with BSE. Several macaques were euthanized at regular intervals starting at 1 year postinoculation, and others were observed until clinical signs developed. Among those who received ≥5 g BSE-inducing agent, attack rates were 100% and prions could be detected in peripheral tissues from 1 year postinoculation onward. The overall median incubation time was 4.6 years (3.7–5.3). However, for 3 macaques orally exposed on multiple occasions, incubation periods were at least 7–10 years. Before clinical signs were noted, we detected a non-type 2B signature, indicating the existence of atypical prion protein during the incubation period. This finding could affect diagnosis of variant Creutzfeldt-Jakob disease in humans and might be relevant for retrospective studies of positive tonsillectomy or appendectomy specimens because time of infection is unknown.
Variant Creutzfeldt-Jakob disease (vCJD) (1) is most likely caused by dietary exposure to bovine spongiform encephalopathy (BSE) prions (2–4). In the United Kingdom, risk for infection with BSE has been considerable, but only 172 cases of vCJD have been documented (5–7). However, the infective dose for oral transmission of the BSE agent to humans is unknown, and incubation times can only be estimated (5,6). In 2001, the European Union funded a risk assessment study in nonhuman primates to estimate the risk for humans exposed to BSE-contaminated food or blood products (8).
A determining factor for susceptibility to BSE prions is a polymorphism for methionine (M) or valine (V) at codon 129 of the human prion protein gene (PRNP). All vCJD cases examined were methionine homozygotes at PRNP codon 129 (129-MM) (9). The overall distribution of PRNP codon 129 genotypes in the general UK population is ≈39% MM, ≈50% MV, and ≈11% VV (7,10). Evidently, persons with a 129-VV genotype can be infected (11), and clinical signs develop after a longer incubation time among those with a 129-MV genotype than among those with a 129-MM genotype (12). However, retrospective analyses of biopsy samples suggest that prevalence of BSE infection is higher among persons who belong to a certain birth cohort and lived in the United Kingdom from 1980 through 1989 (109 cases/million persons [13] to 237 cases/million persons [14]). The reason for the discrepancy between the low number of vCJD cases and higher prevalence of infected persons in the United Kingdom is not known, but the PRNP polymorphism might contribute to this discrepancy as just described. Intriguingly, among hamsters that were orally exposed multiple times to central nervous system (CNS) tissues infected with the scrapie agent, incubation times were significantly prolonged (13). Thus, not only the PRNP polymorphism and the dose but also the mode of transmission might contribute to the development of subclinical cases. However, estimating exposure risks for humans based solely on these results is difficult because of the digestive physiology, life expectancy, and other metabolic parameters of hamsters.
Figure 1

Figure 1. . Schematic diagram of the mature nonglycosylated prion protein and below amino acid sequences of the human and the simian prion polypeptide chain. Homology (198/207 aa) between human and simian mature...
In prion diseases such as CJD, kuru, BSE, scrapie, and chronic wasting disease, the cellular form of prion protein (PrPC) is thought to be converted into abnormal PrP (PrPSc) through a posttranslational event. As a result, PrPSc becomes partially resistant to proteases. The misfolded prion protein comprises an N terminal protease-sensitive part followed by a region with variable protease sensitivity and a C-terminal protease-resistant core referred to as PrPres or PrP27–30 (Figure 1). Limited protease exposure of PrPSc in vitro generates nonglycosylated core fragments of 19–21 kDa (14,15), which are used to distinguish 2 major PrPSc types by electrophoresis. Type 1 core protein has an apparent molecular mass of 21 kDa. Its primary cleavage site is at residue 82. Type 2 core protein migrates to the 19-kDa region and has a primary cleavage site at residue 97 (16,17). Both types can coexist in a considerable number of sporadic CJD (18,19) and vCJD cases (14). Subtypes and strains can be further characterized by their so-called glycoform profile because the nonobligatory addition of 1–2 sugar chains results in 3 differently glycosylated isoforms in PrPC and PrPSc (non-, mono-, and diglycosylated molecules, referred to as a PrPres triplet). In sporadic CJD type 2, the monoglycosylated isotype predominates and is referred to as a type 2A signature; whereas, in vCJD, the diglycosylated isoform predominates (1) and is referred to as a type 2B signature (14,16,19). Additional PrPres fragments have been described, for example the so-called C-terminal fragments of 12/13 kDa (20) and 17 kD (21).
Results of the European Union–funded nonhuman primate risk assessment study, designed to determine the dose at which 50% of macaques will be infected (8), show that a 5-g dose given on 1 occasion infected all macaques. Moreover, multiple exposures to high doses might prolong incubation time. Intriguingly, a non–type 2B PrPres pattern in CNS tissues of macaques during the preclinical phase indicated the existence of an intermediate prion isoform. This finding might be relevant for retrospective studies of tonsillectomy or appendectomy specimens, because the time point of infection in humans with PrPres-positive biopsy specimens is not known. As part of the European Union–funded study, we aimed to determine attack rates and incubation times after oral exposure to 5 g or 16 g of BSE-infected brain material in adult cynomolgus monkeys (Macaca fascicularis).
We orally exposed 18 macaques, each 5 years of age, to brain material from cattle with BSE: 15 macaques on 1 occasion and 3 macaques on multiple occasions. Most animals were kept at the primate center of the Paul-Ehrlich-Institut under biosafety level 3 conditions; the others (macaques M3–M8) were kept at the Swedish Institute for Infectious Disease Control, Stockholm. The study was approved by the Hessian Animal Protection Committee (local permit no. F107/45 and F107/63) and supervised by local authorities (Regierungspräsidium Darmstadt).
The BSE inoculum was a pool of homogenized bovine brain from 11 cows with natural BSE infection (22). The level of infectivity was determined (data not shown) in BoTg110 mice (23). Cynomolgus monkeys were purchased from the Centre de Recherche en Primatologie, Mauritius. All animals were homozygous for M at codon 129 of the PRNP gene (4,22). We fed (in muesli balls, monitored by video to ensure that the total amount was eaten) 5 g of BSE inoculum to each of 12 macaques (macaques S1–S5 and S9–S15), 8–16 g of BSE inoculum to each of 6 macaques (macaques S6–S8 and C1–C3), and mock inoculum (non-BSE–infected brain material) to each of 8 macaques (macaques M1–M8). We had originally planned to inoculate all macaques on 1 occasion; however, because of feeding problems, 3 macaques (C1–C3) had to be inoculated on several occasions. Of these 3 macaques, 1 received a cumulative dose of 8 g, the second 10 g, and the third 16 g (Table 1).
All animals were observed daily for any abnormalities. Cerebrospinal fluid (CSF) samples were collected at regular intervals and examined for biomarkers of brain damage by 14–3-3 protein (14–3-3p) tests (22). We planned 2 studies. For study 1, BSE-infected macaques (groups I and III) were to be kept until development of clinical signs to determine incubation periods. For study 2, macaques (group II) received 5 g BSE brain material on 1 occasion and were euthanized at regular intervals during the incubation period (macaques S9–S15, Table 2). However, study 1 was possible only for macaques S1–S8 (group I, Table 2) because among macaques C1–C3 (group III, Table 2), non–BSE-associated disease necessitated euthanasia. Macaques M1–M8 were the control macaques (group IV) inoculated with non-BSE brain material (Table 2).
During postmortem examinations, brain, spinal cord, gut-associated lymphoid tissues (GALT), lymph nodes, tonsils, and other organs and tissues were either fixed in buffered formaldehyde solution (4% wt/vol) or stored at −80°C as described (22). Routine histopathologic examinations of the brains were performed to detect spongiform lesions in hematoxylin and eosin–stained tissue sections. To detect proteinase K (PK)–resistant PrP fragments in tissue sections, we conducted paraffin-embedded tissue blot analyses (24). Western immunoblot analyses to localize PrPres in homogenized and PK–treated (20 µg PK/mL buffer) tissue samples (50 μg of tissue proteins were loaded onto a lane) were conducted as described (22). Western blot–negative results were retested by using Amersham Hyperfilm ECL (GE Healthcare, Life Sciences Europe, Freiburg, Germany) for visualization. Monoclonal antibodies and polyclonal antiserum were used for immunodetection (Table 3) and epitope mapping of PrPC and PrPSc (Figure 1). Bioassay studies were conducted in BoTg110 mice expressing the bovine PRNP gene (23).
Figure 2

Figure 2. . Percentage macaques surviving after oral inoculation brain material with or without (mock) bovine spongiform encephalopathy (BSE)–-inducing agent. Macaques exposed to 5 g (gray circles) or 16 g BSE (black circles)...
Figure 3

Figure 3. . Western immunoblot analysis of proteinease-resistant prion protein (PrPres) (from brain). The size of the nonglycosylated band was either 21 kDa (termed type 1, sCJD case BN141) or 19 kDa (termed...
Figure 4

Figure 4. . Western blot analysis of bovine spongiform encephalopathy proteinease-resistant prion protein (PrPres) (lumbar spinal cord segments) in preclinical and clinically ill macaques. An atypical PrPres pattern was detectable in macaques euthanized...
Figure 5

Figure 5. . A) Epitope mapping of proteinease-resistant prion protein (PrPres) by Western immunblot analyses (thalamus) from a macaque showing neurologic signs. The PK-sensitive N terminal fragment (mAb 5G5) and the adjacent region...
Among single-dosed macaques, gait ataxia developed in S1–S8, and CSF samples were positive for 14–3-3p from 3.7 through 5.3 years postinoculation; no differences in incubation periods were noted for macaques that received 5 g or 16 g of BSE inoculum (Table 2, Figure 2). Postmortem examinations of all macaques showing neurologic signs detected a type 2B PrPres signature in different brain areas (obex region, cerebellum/deep nuclei, pons, thalamus, caudate nucleus, cortex cerebri) and in all spinal cord segments examined (C1–L4) (Table 2, Figures 3, 4). The type 1–specific monoclonal antibody 12B2 could not detect any PrPres triplets in these specimens (Figure 5, panel A). Typical spongiform changes were seen in hemotoxylin and eosin–stained brain tissue sections (data not shown).
Figure 6
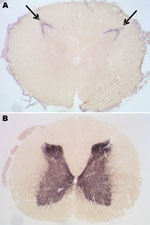
Figure 6. . Paraffin-embedded tissue blot analyses of lumbar spinal cord segments from the preclinical macaque S14 (A) and a clinically ill macaque (B) for detection and localization of proteinease-resistant prion protein (PrPres)...
Among macaques that received 8–16 g of BSE inoculum (C1–C3), no behavioral changes, gait ataxia, or 14–3-3p-positive CSF were detected. However, as they got older, obesity and chronically elevated blood glucose concentrations (>126 mg/dL) developed and were followed by a rapid decrease in body weight. For humane reasons, these animals were euthanized at 6.5 years (C1, C2) and 8.8 years (C3) postinoculation. Retrospective analyses of plasma samples detected normal insulin levels at 3–4 years of age, followed by hyperinsulinemia and a progressive decline in plasma insulin levels. Postmortem examinations of pancreatic tissue sections indicated replacement of the normal islet architecture by islet-associated amyloid and marked reduction of α- and β-cell mass (data not shown). On the basis of the typical changes during the prediabetic phase, clinical parameters, and pancreas histopathology, type 2 diabetes (a common disease for nonhuman primates in captivity) was diagnosed. We detected neither spongiform changes nor PrPres deposits in the brains of these 3 macaques (Table 2). However, an atypical PrPres pattern, a 26-kDa fragment, and a 17-kDa fragment were found in lumbar, but not other, spinal cord segments in 2 of the 3 macaques (Table 2, Figure 4). Paraffin-embedded tissue blot analyses confirmed Western immunoblot results (Figure 6). Paraffin-embedded tissue blot analyses of lymphoid tissues (GALT, lymph nodes) revealed PrPres deposits in 3 macaques (Table 2).
From 3 years postinoculation onward, we also detected an atypical PrPres pattern in lumbar spinal cord segments in 4 of 6 macaques that had received a single 5-g dose of BSE inoculum (macaques S11, S12, S14, S15; Table 2, Figure 4). The lumbar part of the spinal cord is probably the primary site of prion entry into the simian CNS after oral uptake of the BSE agent (data not shown). We did not detect histopathologic changes in hemotoxylin and eosin–stained brain tissue sections from macaques before clinical signs developed (S9–S15, C1–C3).
The atypical molecular signature found in lumbar segments of macaques with subclinical infection was characterized by the predominance of a PrPres fragment, which migrated at the 17-kDa region of the sodium dodecyl sulfate polyacrylamide gel. A second PrPres molecule migrated at the area of 26 kDa but was sometimes hardly visible (Figure 4). Epitope mapping was conducted to characterize these 2 PrP molecules. Paradoxically, all antibodies, including monoclonal antibody 12B2 (Figure 5, panel B), and polyclonal antiserum recognizing the C-terminus (data not shown) could bind to both fragments. Non-PrP antibodies did not bind to the atypical fragments (data not shown). Deglycosylation of PK–treated lumbar spinal cord samples by using peptide -N-glycosidase F treatment resulted in a single band with a molecular weight of ≈22–23 kDa in animals with preclinical/subclinical infection compared with a single 19–20 kDa band in macaques showing neurologic signs (data not shown).
Figure 7

Figure 7. . A) Summary of studies of BSE transmission to macaques and subsequent passage of lumbar spinal cord tissues (L2–L3) from a symptomatic and a preclinical macaque to BoTg110 mice. Inoculation of...
The reason for the abnormal migration behavior of the atypical fragments remains to be determined. When lumbar spinal cord tissue homogenates were intracerebrally inoculated into mice transgenic for the bovine PRNP gene, samples from symptomatic (type 2B signature) and asymptomatic macaques (abnormal signature) were infectious and caused disease in mice (53% diseased mice in both groups) with no statistically significant difference in the incubation periods (355 ± 41 vs. 372 ± 7 days postinoculation, respectively, Plogrank test not significant) (Figure 7). The percentage of mice inoculated with type 2B or non–type 2B material that showed clinical signs was low, probably because of the small amount of gray matter (PrPres) in the inoculum; attack rates were 100% among mice inoculated with gray matter from brain samples (data not shown). Among mice infected with tissue samples from the asymptomatic animals, a molecular signature that differed from that of the inoculum developed, whereas the type 2B signature found in macaques showing neurologic signs was stable after transmission to mice (Figure 7).
Because only a few macaques have died, it will take longer than previously estimated before all data from low-dose (0.05 and 0.005 g) exposures will be available (data not shown). Although all macaques were originally to be inoculated with BSE-infected cattle brain material on only 1 occasion, 3 of 6 macaques receiving >5 g of inoculum had to be fed on multiple occasions.
The attack rate after dietary exposure to ≥5 g BSE brain material in 5-year-old adult 129-MM cynomolgus monkeys was 100% (18/18). PrPres deposits could be detected outside the GALT in gut-draining lymph nodes from at least 1 year postinoculation onward. These data show that the dose at which 50% of these nonhuman primates will be infected will be distinctly lower than previously estimated (4).
For single-dosed animals, the incubation period was 4.6 years (median, range 3.7–5.3 years). There was no difference between those that received 5 g and 16 g, indicating that <5 g represented the dose at which disease developed in 100% of macaques (LD100). The shortest incubation period was detected in macaque S6, which was given 16 g of inoculum on 1 occasion. This short incubation period might have resulted from the extremely high dose. However, retrospective analyses revealed that macaque S6 had a highly stimulated immune system (data not shown), which might also explain the short incubation period. The low variability of incubation periods (4.3–5.3 years, excluding 3.7 years for macaque S6) was probably the result of the high dose. When we inoculated macaques with lower doses (data not shown) or when macaques were inoculated on multiple occasions, incubation periods were highly variable. After the macaques received multiple oral doses, clinical signs of a spongiform disease had not developed by 6.5 (2 of 3 macaques) to 8.8 years (1 of 3 macaques) postinoculation although they received an LD100 on day 1 (Table 1).
Unfortunately, type 2 diabetes developed in all 3 of these macaques as they aged, and they had to be euthanized for humane reasons at the indicated time points. At postmortem examinations, lumbar spinal cord segments were PrPres positive for the 2 macaques (C1 and C2) euthanized 6.5 years postinoculation. We estimate that incubation periods in these 2 animals must be at least 7 years because it took ≥6 months until PrPres deposits were also detectable in the cerebellum/cortex cerebri, thereby causing clinical signs (data not shown). In the third macaque (C3) euthanized 8.8 years postinoculation with a cumulative dose of 16 g, PrPres deposits could only be detected outside the CNS, thereby indicating an estimated incubation period >10 years. Similar results have been described for hamsters orally infected with the scrapie strain 263K on a single or multiple occasions. In that study, a cumulative dose significantly prolonged incubation periods, although hamsters were given much lower doses than were the macaques (13). The upper reference margin using 3× the standard deviation (3σ) of animal group I is 1.52 years, corresponding to a calculated upper limit of the incubation period of 6.1 years after a single high-dose exposure (5–16 g each). This calculated incubation period is distinctly lower than the estimated incubation time of >7–10 years within animals of group III, indicating a biological effect of the successive BSE challenge mode on the incubation time in the macaque model.
The discrepancy between the low number of vCJD cases in the United Kingdom to date and the higher prevalence of infected humans estimated on the basis of retrospective biopsy analyses (27,28) indicates the existence of pre- or subclinical cases, perhaps as a result of a low-dose exposure to BSE-contaminated material or a less susceptible PRNP genotype. We showed that multiple exposures to high doses of BSE prolonged incubation periods in a nonhuman primate model. These findings show that a successive BSE challenge mode might contribute to the development of pre- or subclinical cases despite a susceptible PRNP phenotype and an LD100. This finding is relevant because it is quite likely that most of the UK population has been exposed to BSE-contaminated food on multiple occasions (5,6).
The underlying mechanism of a prolonged incubation period after multiple exposures to an agent that induces a transmissible spongiform encephalopathy is not known (6,13). Theoretically, interference between types or strains could have caused this phenomenon, as has been shown by others (14,19,29,30). However, Diringer et al. (13) used 1 well-defined laboratory scrapie strain (263K) that could also cause prolonged incubation periods in Syrian hamsters after multiple oral exposures. Their finding shows that at least 1 other not-yet identified mechanism causes prolonged incubation periods after multiple oral exposures to agents that induce transmissible spongiform encephalopathy.
Unexpectedly, we detected a non–type 2B PrPres pattern in preclinical cases from 3 years postinoculation onward. Transmission studies in BoTg110 mice showed that tissues were infectious but that this atypical molecular signature was not stable after the first passage to transgenic mice carrying the bovine PrP gene (Figure 7). However, the PK-sensitive N terminal part, the variable region of PK, and the C-terminal end were detectable in both atypical PrP molecules by epitope mapping studies. Thus, at least the 17-kDa molecule showed migration behavior on sodium dodecyl sulfate polyacrylamide gel electrophoresis, which did not correlate with its formal molecular weight. Posttranslational modifications can cause a gel-shifting phenomenon (i.e., anomalous gel mobility), as observed for the phosphorylated tau protein (31). However, it remains to be determined which mechanism caused this anomalous gel mobility. This atypical signature probably reflects neither types nor strains but rather an intermediate conformation of the pathologic PrP.
In conclusion, the LD100 of brain from BSE-infected cattle for 129-MM 5-year-old adult macaques exposed on 1 occasion is <5 g. However, this dose did not cause disease within a prolonged incubation time when animals were exposed on multiple occasions. This finding may be relevant for modeling exposure risks for foodborne prion diseases including chronic wasting disease (32). Moreover, the time-dependent shift of the molecular signature might be relevant for retrospective analyses of biopsy samples most likely from animals with pre- or subclinical vCJD.
Dr Holznagel is a senior staff scientist at the Paul-Ehrlich-Institut. He performed all animal experiments and designed and supervised all laboratory work for this study.
Acknowledgments
We are indebted to Anatoli Rempel, Jelica Cabraja, Martin Stellwagen, Christin Stellwagen, and Viola Jakob for excellent animal husbandry and assistance.
This research was supported by grants of the European Community (BMH4-CT98-6029, QLK1-2002-01096) and by the German Ministry of Health, Bonn/Berlin.
References
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–5. DOIPubMedGoogle Scholar
- Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–90. DOIPubMedGoogle Scholar
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. DOIPubMedGoogle Scholar
- Lasmézas CI, Comoy E, Hawkins S, Herzog C, Mouthon F, Konold T, Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet. 2005;365:781–3 .PubMedGoogle Scholar
- Valleron AJ, Boelle PY, Will R, Cesbron J-Y. Estimation of epidemic size and incubation time based on age characteristics of vCJD in the United Kingdom. Science. 2001;294:1726–8. DOIPubMedGoogle Scholar
- Gravenor MB, Stallard N, Curnow R, McLean AR. Repeated challenge with prion disease: the risk of infection and impact on incubation period. Proc Natl Acad Sci U S A. 2003;100:10960–5. DOIPubMedGoogle Scholar
- Andrews NJ. Incidence of variant Creutzfeldt-Jakob disease diagnoses and death in the U.K. 2011. The National Creutzfeldt-Jakob Disease Research & Surveillance Unit [cited 2013 Feb 28]. http://www.cjd.ed.ac.uk/documents/cjdq72.pdf
- Hunsmann G. Hahmann U, Motzkus D, Bierke P, Dormont D, Lazmezas CI, et al. BSE transmission through food and blood products: a study in primates to assess the risk for humans. 5th Framework Programme [cited 2013 Feb 28].
ftp://ftp.cordis.europa.eu/pub/fp7/kbbe/docs/tse_ok-web.pdf - Mead S, Poulter M, Uphill J, Beck J, Whitfield J, Webb TEF, Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol. 2009;8:57–66. DOIPubMedGoogle Scholar
- Mercier G, Dieterlen F, Lucotte G. Population distribution of the methionine allele at the PRNP codon 129 polymorphism in Europe and the Middle East. Hum Biol. 2008;80:181–90. DOIPubMedGoogle Scholar
- Ironside JW, Bishop MT, Connolly K, Hegazy D, Lowrie S, Le Grice M, Variant Creutzfeldt-Jakob disease: prion protein genotype analysis of positive appendix tissue samples from a retrospective prevalence study. BMJ. 2006;332:1186–8. DOIPubMedGoogle Scholar
- Kaski D, Mead S, Hyare H, Cooper S, Jampana R, Overell J, Variant CJD in an individual heterozygous for PRNP codon 129. Lancet. 2009;374:2128. DOIPubMedGoogle Scholar
- Diringer H, Roehmel J, Beekes M. Effect of repeated oral infection of hamsters with scrapie. J Gen Virol. 1998;79:609–12 .PubMedGoogle Scholar
- Yull HM, Ritchie DL, Langeveld JPM, van Zijderveld FG, Bruce ME, Ironside JW, Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:151–7. DOIPubMedGoogle Scholar
- Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A. 2000;97:10168–72. DOIPubMedGoogle Scholar
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33. DOIPubMedGoogle Scholar
- Cali I, Castellani R, Yuan J, Al-Shekhlee A, Cohen ML, Xiao X, Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain. 2006;129:2266–77. DOIPubMedGoogle Scholar
- Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A. Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol. 2005;4:805–14. DOIPubMedGoogle Scholar
- Cali I, Castellani R, Alshekhlee A, Cohen Y, Blevins J, Yuan J, Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain. 2009;132:2643–58. DOIPubMedGoogle Scholar
- Zou W-Q, Capellari S, Parchi P, Sy M-S, Gambetti P, Chen SG. Identification of novel proteinase K–resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem. 2003;278:40429–36. DOIPubMedGoogle Scholar
- Notari S, Strammiello R, Capellari S, Giese A, Cescatti M, Grassi J, Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem. 2008;283:30557–65. DOIPubMedGoogle Scholar
- Yutzy B, Holznagel E, Coulibaly C, Stuke A, Hahmann U, Deslys J-P, Time-course studies of 14–3-3 protein isoforms in cerebrospinal fluid and brain of primates after oral or intracerebral infection with bovine spongiform encephalopathy agent. J Gen Virol. 2007;88:3469–78. DOIPubMedGoogle Scholar
- Castilla J, Gutierrez A, Pintado B, Ramirez MA, Parra B, Doyle D, Early detection of PrPres in BSE-infected bovine transgenic mice. Arch Virol. 2003;148:677–91 . DOIPubMedGoogle Scholar
- Schulz-Schaeffer WJ, Tschoke S, Kranefuss N, Drose W, Hause-Reitner D, Giese A, The paraffin-embedded tissue blot detects PrP(Sc) early in the incubation time in prion diseases. Am J Pathol. 2000;156:51–6. DOIPubMedGoogle Scholar
- Holznagel E, Yutzy B, Schulz-Schaeffer WJ, Hanschman K-M, Stuke A, Hahmann U, Increase in CD230 (cellular prion protein) fluorescence on blood lymphocytes in bovine spongiform encephalopathy–infected nonhuman primates. Transfusion. 2010;50:452–66. DOIPubMedGoogle Scholar
- Choi J-K, Park S-J, Jun Y-C, Oh J-M, Jeong B-H, Lee H-P, Generation of monoclonal antibody recognized by the GXXXG motif (glycine zipper) of prion protein. Hybridoma (Larchmt). 2006;25:271–7. DOIPubMedGoogle Scholar
- de Marco MF, Linehan J, Gill ON, Clewley JP, Brandner S. Large-scale immunohistochemical examination for lymphoreticular prion protein in tonsil specimens collected in Britain. J Pathol. 2010;222:380–7. DOIPubMedGoogle Scholar
- Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004;203:733–9. DOIPubMedGoogle Scholar
- Nishida N, Katamine S, Manuelidis L. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science. 2005;310:493–6. DOIPubMedGoogle Scholar
- Shikiya RA, Ayers JI, Schutt CR, Kincaid AE, Bartz JC. Coinfecting prion strains compete for a limiting cellular resource. J Virol. 2010;84:5706–14. DOIPubMedGoogle Scholar
- Lindwall G, Cole D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301–5 .PubMedGoogle Scholar
- Race B, Mead-White KD, Miller MW, Barbian KD, Rubenstein R, LaFauci G, Susceptibilities of nonhuman primates to chronic wasting disease. Emerg Infect Dis. 2009;15:1366–76. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 19, Number 5—May 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Edgar Holznagel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51–59, 63225 Langen, Germany
Top