Volume 19, Number 12—December 2013
Research
Spontaneous Generation of Infectious Prion Disease in Transgenic Mice
Cite This Article
Citation for Media
Abstract
We generated transgenic mice expressing bovine cellular prion protein (PrPC) with a leucine substitution at codon 113 (113L). This protein is homologous to human protein with mutation 102L, and its genetic link with Gerstmann–Sträussler–Scheinker syndrome has been established. This mutation in bovine PrPC causes a fully penetrant, lethal, spongiform encephalopathy. This genetic disease was transmitted by intracerebral inoculation of brain homogenate from ill mice expressing mutant bovine PrP to mice expressing wild-type bovine PrP, which indicated de novo generation of infectious prions. Our findings demonstrate that a single amino acid change in the PrPC sequence can induce spontaneous generation of an infectious prion disease that differs from all others identified in hosts expressing the same PrPC sequence. These observations support the view that a variety of infectious prion strains might spontaneously emerge in hosts displaying random genetic PrPC mutations.
Transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative diseases that affect humans and animals and involve pathologic conversion of host cellular prion protein (PrPC) to a disease-related isoform (PrPSc), as proposed in the protein-only hypothesis (1). Depending on how these encephalopathies originate, TSEs are classified as sporadic, genetic, or infectious. Most have been experimentally transmitted and, with some exceptions, the presence of PrP resistant to proteinase K digestion (PrPres) is related to their infectivity (2–4).
Human genetic TSEs are caused by >30 autosomal-dominant point mutations in the human prion protein gene (Prnp) and have been classified as Gerstmann–Sträussler–Scheinker syndrome, familial Creutzfeldt–Jakob disease, or fatal familial insomnia (FFI), according to the clinical symptoms. Some of these genetic diseases have been transmitted to primates or rodents, although transmission rates were low in most instances (5–8). Regarding TSEs, pathogenic mutations in Prnp are believed to predispose mutant PrPC to convert spontaneously to a pathogenic isoform (9–11).
Several transgenic mouse models confirmed that PrPC with mutations induces a spectrum of neurologic diseases with clinical or histologic features of TSEs (12–15). However, the crucial prediction that a disease-associated PrP mutation can spontaneously generate infectivity has only been demonstrated in mice carrying the mutation D177N, the mouse equivalent of the mutation associated with human FFI (16). Spontaneous appearance of infectivity has also been reported in transgenic mice expressing a mouse PrPC with 2 point mutations (170N and 174T) that subtly affect the structure of its globular domain (17).
The first described and most common Gerstmann–Sträussler–Scheinker syndrome mutation causing ataxia is P102L (18,19). Bovine P113L, which has a leucine substitution at codon 113, is homologous to human P102L and mouse P101L. Although bovine PrPC with the 113L mutation has not been found in nature, it would be useful to establish whether this mutation could induce spontaneous generation of an infectious prion disease in a bovine PrP context. In this study, we analyzed the phenotype of transgenic mice expressing mutant 113L bovine prion protein (BoPrP) and the ability of these mice to generate de novo infectious prions in comparison with control mice expressing the wild-type protein.
Ethics
Animal experiments were conducted in strict accordance with recommendations in the guidelines of the Code for Methods and Welfare Considerations in Behavioral Research with Animals (Directive 86/609EC). All efforts were made to minimize detrimental effects on animals. Experiments were approved by the Committee on the Ethics of Animal Experiments of the INIA Institute (permit no. CEEA2009/004).
Transgenic Mice
The open reading frame (ORF) of the bovine Prnp gene was isolated by PCR amplification and cloned in a pGEM-T vector as described (20). The ORF was mutated by using the QuikChange II-XL Kit (Stratagene, La Jolla, CA, USA) with specific oligonucleotides (5′-CGGTCAGTGGAACAAGCTCAGTAAGCCGAAAACC-3′ and 5′-GGTTTTCGGCTTACTGAGCTTGTTCCACTGACCG-3′) according to procedures of the manufacturer. The P113L-PrP ORF was excised from the cloning vector by using restriction enzyme XhoI and inserted into MoPrP.Xho vector (21), which was also digested with XhoI. This vector contains the murine PrP promoter and exon-1, intron-1, exon-2, and 3′-untranslated sequences. Transgenic mice were generated by microinjection of DNA according to a published procedure (22).
Neuropathologic Studies in Spontaneously Diseased 113LBoPrP-Tg Mice
Brains were rapidly harvested from the skulls and fixed in 4% paraformaldehyde in phosphate buffer. Coronal slabs were embedded in paraffin and 5-μm sections of cerebrum, cerebellum, brain stem, and spinal cord were obtained by using a sliding microtome. De-waxed sections were stained with hemotoxylin and eosin, Congo red, or thioflavin, or processed for immunohistochemical analysis. Immunohistochemical analysis for detection of glial fibrillary acidic protein (GFAP) and cleaved caspase-3 was conducted by using a modified labeled streptavidin technique (LSAB2-System peroxidase; Dako, Glostrup, Denmark). The rabbit polyclonal antibody to GFAP (Dako) was used at a dilution of 1:600. Cleaved caspase-3 rabbit polyclonal antibody (D175, cell signaling) was used at a dilution of 1:50. Microglial cells were stained with the biotinylated lectin from Lycopericon esculentum (L-0651; Sigma, St. Louis, MO, USA) and used at a dilution of 1:100. PrP was immunolabeled with monoclonal antibody (mAb) 6H4 (Prionics, Schlieren, Switzerland) and used at a dilution of 1:30 in sections pretreated with 35% HCl for 2 min at 100°C and then with 96% formic acid for 10 min at room temperature. These procedures have been detailed elsewhere (23).
Mouse Transmission Studies
For transmission studies, we used the Tg110 mouse line (20) that expresses wild-type bovine-PrPC in a mouse Prnp null background. Inocula were prepared from brain tissues as 10% (wt/vol) homogenates. Individually identified 6–10-week-old mice were anesthetized and inoculated with 2 mg of 10% brain homogenate in the right parietal lobe by using a 25-gauge disposable hypodermic needle. Mice were observed daily and neurologic status was assessed weekly. When progression of TSE was evident or evident at 650 days postinoculation (dpi), animals were euthanized for ethical reasons. Once mice were euthanized, brains were collected, frozen, and analyzed by Western blotting. Samples fixed in buffered 10% formalin underwent histologic analysis, immunohistochemical analysis, or histoblotting. Spleens were frozen for Western blot analysis.
Western Blot Analyses of PrPres
Frozen mouse brain samples were prepared as 10% (wt/vol) homogenates in 5% glucose in distilled water in grinding tubes (Bio-Rad, Hercules, CA, USA) by using a TeSeE Precess 48 homogenizer (Bio-Rad) following the manufacturer’s instructions. Samples were analyzed by Western blotting as described (24). For immunoblotting experiments, mAbs Sha31 (25), 9A2 (26), 12B2 (26), Saf84 (25), and R145 (Vetyerinary Laboratories Agency, Weybridge, UK) were used at concentrations of 1 μg/mL. Sha31 recognizes 156YEDRYYRE163 epitope, 9A2 recognizes 110WNK112 epitope, 12B2 recognizes 101WGQGG105 epitope, Saf84 recognizes 175RPVDQY180 epitope, and R145 recognizes 231RESQUA235 epitope of the bovine PrP sequence.
Histopathologic Analysis
All procedures involving brains from infected mice were performed as described (27). Samples were fixed in neutral-buffered 10% formalin (4% formaldehyde) before being embedded in paraffin. Once deparaffinated, 2-µm tissue sections were stained with hematoxylin and eosin. Lesion profiles of brains were established according to the standard method described by Fraser and Dickinson (28). For paraffin-embedded tissue blots, the protocol described by Andréoletti et al. (29) was used.
Expression of 113LBoPrP in Transgenic Mice
Figure 1
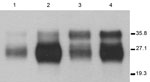
Figure 1. . . Immunoblots of brain extracts from hemizygous 113LBoPrP-Tg037+/− 113LBoPrP-Tg009+/− mouse lines compared with those of cow brain extract and BoPrP-Tg110 mouse brain extract. Brain homogenates were analyzed by Western blotting...
Seven lines (founders) of 113LBoPrPC heterozygous transgenic mice carrying the endogenous murine Prnp gene (Prnp mu+/− 113Lbo+/−) were obtained. Lines 113LBoPrP-Tg037 and 113LBoPrP-Tg009 were selected on the basis of their expression levels, and bred to homozygosity in a murine Prnp null background. To achieve this expression, selected lines were crossed with Prnp null mice (Prnp mu−/−) to achieve transgene-hemizygous lines (Prnp mu−/− 113Lbo+/−). Absence of the murine Prnp gene was determined by using PCR with specific primers. Transgene expression levels were then determined in brain homogenates by serial dilution and compared with PrPC levels found in bovine brain homogenates. Transgene expression levels for the two 1-month-old mice with hemizygous Tg lines 113LBoPrP-Tg037 and 113LBoPrP-Tg009 were found to be ≈3× and 0.5×, respectively. Mutant 113LBoPrP expressed in 009 and 037 transgenic lines showed an electrophoretic profile similar to that of wild-type bovine PrPC from BoPrP-Tg110 mice or cow brain, although only small differences in glycoform ratios were observed (Figure 1). Next, by crossing hemizygous animals, we obtained transgene-homozygous animals (Prnp mu−/− 113Lbo+/+) (30).
Spontaneous Neurologic Disease in Transgenic Mice Expressing Mutant 113LBoPrP
Spontaneous neurologic disease developed in 113LBoPrP-Tg037 mice expressing mutant 113LBoPrP. These mice had reduced lifespans compared with either non-Tg (Prnp−/−) mice or transgenic mice expressing similar or higher levels of wild-type BoPrP (Table 1). However, disease did not develop in 113LBoPrP-Tg009 mice expressing low levels of mutant protein, and these mice had survival times similar to non-Tg (Prnp−/−) mice or BoPrP-Tg110 mice. Onset of clinical signs and survival times were dependent on the expression level of 113LBoPrP (Table 1) (i.e., transgene-homozygous 113LBoPrP-Tg037 mice showed an earlier onset of clinical signs and reduced survival times than hemizygous mice of the same line). Neurologic alterations generally involved motor impairment with ataxia affecting mainly the hind limbs. Mice showed a rough coat and prominent hunch at the early stages of clinical signs. Most mice had a wobbling gait and slight paralysis in the back limbs. Some mice had conjunctivitis and showed compulsive scratching in the head area. At the end stage of the disease, mice had highly restricted movement and lethargy. No signs of hyperactivity were detected in these mice.
Neuropathologic Alterations in Transgenic Mice Expressing Mutant 113LBoPrP
Figure 2
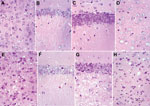
Figure 2. . . Comparison between homozygous bovine prion protein (BoPrP)-Tg110+/+ control mice (panels A–D) and hemizygous 113LBoPrP-Tg037+/− mice with end-stage disease (panels E–H) in parietal cortex (panels A and E), CA1 region...
All 113LBoPrP-Tg037 mice at the terminal disease stage showed spongiosis in the cerebral cortex, thalamus, and hilus of the dentate gyrus, but not in the CA1 region of the hippocampus and granule cell layer of the dentate gyrus, compared with age-matched control BoPrP-Tg110 mice (Figure 2). Marked granule cell loss, spongiosis in the molecular layer, granule cell layer, subcortical white matter, and Bergmann glia hypertrophy and hyperplasia were also observed in these mice at the terminal disease stage. However, at the early stages of clinical signs, no spongiform changes were found, although neuronal loss was observed when 113LBoPrP-Tg037 mice were compared with age-matched control BoPrP-Tg110 mice. These findings were particularly evident in the hippocampus proper (including hilus) and granular cell layer of the cerebellum.
Changes were more pronounced in animals with severe clinical manifestations. Neurons with a shrunken cytoplasm and nucleus were observed in all 113LBoPrP-Tg037 mice, and these appeared in the molecular layer of the cerebellum, hippocampus (mainly plexiform layers and hilus), thalamus, and pons. Morphologic PrP aggregates, congophilic materials, or thioflavin-positive deposits were not detected in 113LBoPrP-Tg037 mice. Astrocyte gliosis was observed throughout the brain in 113LBoPrP-Tg037 mice, even at the early stage of the disease. The number and size of reactive astrocytes increased, as shown by immunolabeling with antibody against GFAP, in the cerebral cortex, hippocampus, striatum, thalamus, cerebellum, and brain stem. Microglial proliferation, as visualized with Lycopericum esculentum lectin, was evident in brains of 113LBoPrP-Tg037 mice but not in age-matched control mice.
Modification of Biochemical Properties of Bovine-PrPC by Mutation 113L
Figure 3
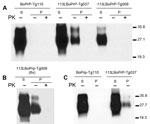
Figure 3. . . Host cellular prion protein (PrPC) solubility and proteinase K (PK) resistance studies in homozygous 113LBoPrP-Tg037, 113LBoPrP-Tg009, and control BoPrP-Tg110 mice. Western blot analysis with monoclonal antibody 2A11 of soluble...
To explore changes in biochemical properties of mutant 113LBoPrP, we solubilized brain homogenates from control BoPrP-Tg110, 113LBoPrP-Tg037, and 113LBoPrP-Tg009 mouse lines in extraction buffer and ultracentrifuged them at 100,000 × g for 1 h. Western blotting of soluble and insoluble fractions indicated differential biochemical properties of mutant 113LBoPrP compared with wild-type BoPrP (Figure 3); the 113L mutation resulted in a more insoluble protein. This insolubility was detected in 113LBoPrP-Tg037 and 113LBoPrP-Tg009 mouse lines. However, when the 113LBoPrP-Tg009 mouse line was compared with the other 2 mouse lines, the expression level was lower and insolubility was detectable only when an 8-fold equivalent brain tissue mass was used (Figure 3, panel B) to obtain equivalent PrP signal.
Insolubility was detected early in the lifespan (30 days after birth) of mice (Figure 3, panel C), which suggested that quantification of 113LBoPrP would reflect a cumulative effect. PK resistance was not found in mutant 113LBoPrP or in wild-type BoPrP, which were digested at the PK concentration used (Figure 3).
Spontaneous Generation of Infectious Prions by 113LBoPrP-Tg037 Mice
Figure 4
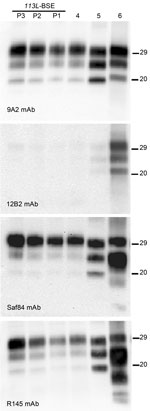
Figure 4. . . Comparative Western blot analyses of brain prion protein resistant to proteinase K digestion (PrPres) from BoPrP-Tg110 mice infected with bovine spongiform encephalopathy (BSE)-C, 113L-BSE, BSE-L, and BSE-H prions. Mice...
To test potential infectivity of brains of mutant 113LBoPrP-Tg037 mice, we intracerebrally inoculated brain homogenates from sick animals into transgenic mice expressing wild-type bovine PrP (BoPrP-Tg110). In the first experiment (Table 2), we used a brain homogenate from a unique terminally sick animal. In this instance, neurologic signs developed in only 1 of 5 (attack rate 20%) inoculated Tg110 mice. This mouse died at 333 dpi and contained a considerable amount of PrPres in its brain, as shown by Western blot (Figure 4).
When brain homogenate from this mouse was reinoculated into 6 Tg110 mice (second passage), neurologic signs developed in all recipients, and these mice showed a shorter mean ± SEM incubation period (272 ± 38 dpi), which was suggestive of an increased infectious titer that was maintained in subsequent passages (Table 2). These results were confirmed in a second independent experiment with a brain homogenate derived from a pool of 5 terminally sick 113LBoPrP-Tg037 mice. In this instance, 2 of 6 (attack rate 33%) inoculated mice were infected (Tg110 mice) (33% attack rate) and had incubation periods of 322 and 406 dpi, respectively. As in the first experiment, second passage (using a pool containing both mouse brains) produced an attack rate of 100% and a shorter incubation period (291 ± 23 dpi), which were maintained in subsequent passages (Table 2).
Brain homogenate from Tg110 mice expressing comparable amounts of wild-type BoPrP, as well as brain homogenate from healthy mice, was also used to inoculate uninfected mice, which served as negative controls. Neurologic signs did not develop in any of the inoculated mice (Table 2). These mice were euthanized at 650 dpi and did not show any PrPres in their brains by Western blot.
For comparative studies, material from the brainstem of cows that contained classical bovine spongiform encephalopathy (BSE), atypical BSE-H, and atypical BSE-L prions was also inoculated into mice by the same procedure. These 3 inocula induced a typical neurologic disease after primary transmission and showed an attack rate of 100% (Table 2). Survivals times of mice inoculated with brainstem of 113LBoPrP-Tg037 mice on second and third passages were similar to those produced by the classical BSE-C isolate (Table 2), as well as by other isolates reported for the same Tg110 mouse line (20,22,32).
Properties of P113L-BSE Prion
Western blot analysis with mAb 9A2 against brain-PrPres produced by BoPrP-Tg110 mice infected with the new 113L-BSE prion (Figure 4, panel A) showed a typical BSE PrP banding pattern characterized by small fragments (19-kDa fragment for the aglycosyl band) and prominent diglycosylated species in all challenged PrPres-positive mice. This result was indistinguishable from that produced by classical BSE-C prion in these mice but differed from that observed after inoculation with atypical BSE-H or BSE-L prions (Figure 4, panel A).
Further characterization of PrPres with other mAbs showed that the new 113L-BSE prion was not recognized by mAb 12B2 (Figure 4, panel B), whose epitope (101WGQGG105 according to the bovine PrP sequence) is known to be poorly protected from PK digestion (26,32) in the classical BSE-derived prion but well preserved in the atypical BSE-H prion (Figure 4, panel B) (31). Furthermore, PrPres immunolabeling with mAbs Saf84 and R145 showed that mice infected with the new 113L-BSE prion, in contrast to mice infected with the H-type prion, did not show the characteristic PrPres band profile (4 bands) of cattle BSE-H, but showed a PrPres-profile (3 bands) similar to that of the BSE-C prion (Figure 4, panels C, D).
Comparative study of PrPSc accumulation in spleen from Tg110-infected mice showed that mice infected with 113L-BSE or BSE-C prions consistently showed positive results for presence of PrPres by Western blot. In contrast, no PrPSc deposits were detected in mice infected with either BSE-L or BSE-H prions. Similar results were obtained in subsequent passages.
Figure 5
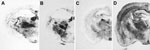
Figure 5. . . Immunochemical analysis of paraffin-embedded tissue blots of representative coronal sections of the hippocampus, showing deposition patterns of abnormal isoform of host-encoded prion protein in brains from BoPrP-Tg110 mice infected...
Figure 6

Figure 6. . . Vacuolar lesion profile in brains from BoPrP-Tg110 mice inoculated with bovine spongiform encephalopathy (BSE)-C (black circles, n = 6 animals), BSE-H (black triangles, n = 6 animals), BSE-L (black...
We next examined vacuolation and PrPSc distribution in the brain, which are known to vary by strains/TSE prions (28,33). In general, we observed that PrPSc deposition patterns in brains of 113L-BSE–infected mice were different from mice infected with BSE-H or BSE-L prions, but these overlapped mostly with those infected with BSE-C prion (Figure 5). However, 113L-BSE–infected mice at the terminal stage of disease showed only spongiform changes that remained limited to the thalamus even after 3 passages (Figure 6). This finding was in contrast with lesion profiles observed in the mice infected with BSE-C, BSE-H or BSE-L prions (Figure 6) in which substantial vacuolar changes were observed in various brain areas. These results indicate that 113L-BSE is an authentic infectious prion that phenotypically differs from BSE-H and BSE-L prions but has biochemical characteristic and histopathologic features similar to those of the classical BSE-C prion.
We showed that the 113L mutation in the bovine Prnp gene gives rise to a spontaneous neurodegenerative disease when expressed in transgenic mice. Neurologic symptoms of ataxia, rigidity, and lethargy accompanied by spongiform degeneration throughout the brain spontaneously develop in these mice. The rate at which illness progresses is related to expression levels of the mutant 113LBoPrP (Table 1). Neurologic alterations did not develop in several mouse lines expressing similar or higher levels of wild-type bovine PrPC during their lifespan, which is similar to observations in wild-type mice (Table 1) (20). Although the mechanism inducing the disease is unclear, we suggest that the 113L mutation in bovine PrPC could give rise to a different structure with respect to wild-type PrPC, which shows reduced solubility (Figure 3). Enhanced aggregation of mutant PrP could affect the appearance of the disease. Other mutations in the Prnp gene have also been related to enhanced aggregation of the mutant PrP in transgenic mice (12–14,34). However, the mechanisms through which these mutations may influence the aggregation properties of PrPC are unclear.
In previous studies, overexpression of murine PrP carrying the 101L mutation (equivalent to human 102L and bovine 113L mutations) led to spontaneous neurodegenerative disease in mice (15,35,36). However, when this mutation was introduced into the murine Prnp gene by gene targeting, mice homozygous for the 101L mutation showed no spontaneous spongiform encephalopathy (37). As proposed by Manson et al., the lifespan of a mouse carrying only 1 or 2 copies of the mutant gene is insufficiently long enough to enable the stochastic event that makes TSE occur (37). Transgenic mice expressing high levels of human PrPC carrying the familial 101L mutation were reported to be free of disease (38). These results suggest that an equivalent mutation in PrPs from different species might have different structural consequences. A possible explanation is that species-specific interaction sites for PrP cofactors or chaperones are required, and that in mice they are compatible for bovine PrP but not for human PrP.
We also show that spontaneous neurodegenerative disease induced by the single 113L amino acid substitution is transmissible to mice expressing wild-type bovine PrPC, indicating spontaneous generation of infectious prions. Transmissibility of this genetically initiated disease to mice not carrying 113L mutations provides crucial support for a causal link between PrP misfolding and the spontaneous generation of a transmissible prion. Whether the small amount of insoluble PrP we detected in brain homogenates (Figure 3) constitutes the infectious prion in our mice, or some other as yet uncharacterized species, remains to be determined.
Several transgenic mouse models expressing PrP with various familial mutations have been reported (12–14,34).Most of these transgenic mouse models have confirmed that the presence of these mutations triggers spontaneous disease, but spontaneous generation of a transmissible prion has only been reported for mutation D178N, associated with human FFI (16). This study reported the spontaneous appearance of infectivity in knock-in mice carrying the mouse-equivalent D177N mutation. Spontaneous infectivity has also been reported in transgenic mice expressing a mouse PrP with 2 point mutations (170N and 174T), which subtly affect the structure of its globular domain (17).
The new 113L BSE prion generated shares some phenotypic features with the classical BSE-C prion when inoculated in the same Tg110 mouse line according to various criteria: 1) apparent molecular mass of PrPres, 2) PrPres glycosylation pattern, 3) lack of immunoreactivity with mAb 12B2, 4) pattern of labeling with mAbs Saf84 and R145, 5) detectable PrPres in spleens of infected animals, and 6) spatial distribution of PrPres in brain. However, the vacuolation profile in brain was distinct from those of all known bovine prion strains (classical BSE-C, atypical BSE-H, and atypical BSE-L prions). These differences were maintained after subsequent passages, indicating that the novel prion, spontaneously produced by transgenic mice expressing mutant 113LBoPrP, is distinct from all known bovine prion strains, although it shares many phenotypic features with the classical BSE-C prion.
These observations demonstrate that mutations in bovine PrP can result in spontaneous generation of infectious prion diseases and support the hypothesis of a genetic origin for the epidemic BSE prion. Different features exhibited by the new 113L-BSE prion, compared with those of the classical BSE prion, suggest that if the origin of BSE was genetic, it is unlikely that the causal mutation would be related to the 113L mutation. However, slight phenotypic differences observed could be the results of evolution of the epidemic BSE prion in field conditions in cattle, which must be different from those of our transgenic mouse model. Although BoPrP with the 113L mutation has not been found in nature, a potential pathogenic mutation (E211K) within PrP has been recently reported in a cow with an H-type BSE phenotype (39). This mutation is equivalent to a common mutation (E200K) in humans, which is associated with genetic TSEs.
Spontaneous appearance of infectivity reported in transgenic mice expressing a mutated BoPrP and in mice expressing mutated mouse PrP reported by Stöhr et al. (40) supports the hypothesis that infectious TSE prions, could originate by a random genetic mutation that can induce de novo generation of infectious prions, and that this mechanism could constitute a source of prion diversity. These considerations enable us to hypothesize that the BSE epidemic could have begun by a random genetic mutation that was able to generate de novo infectious prions, which were included in meat and bone meal fed to cattle and then broadly expanded in the cattle population. According to this hypothesis, a key strategy for controlling BSE would involve preventing cows from consuming products from cows with spontaneous cases of BSE.
Dr Torres is lead researcher scientist in the Prions Group at the Centro de Investigación en Sanidad Animal–Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Madrid, Spain. His research interests include prion strain characterization and evolution and the pathogenesis of prion diseases and their effects on human and animal health.
Acknowledgment
This study was supported by grants from the Spanish Ministerio de Ciencia e Innovación (EET2002-5168 and AGL2009-11553-C02-02) and the European Union (FP7-2009-CT-222887 and FP7-2009-CT-228394).
References
- McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35:57–62 . DOIPubMedGoogle Scholar
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. DOIPubMedGoogle Scholar
- Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–22. DOIPubMedGoogle Scholar
- Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–29. DOIPubMedGoogle Scholar
- Tateishi J, Kitamoto T, Hoque MZ, Furukawa H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology. 1996;46:532–7. DOIPubMedGoogle Scholar
- Mastrianni JA, Capellari S, Telling GC, Han D, Bosque P, Prusiner SB, Inherited prion disease caused by the V210I mutation: transmission to transgenic mice. Neurology. 2001;57:2198–205. DOIPubMedGoogle Scholar
- Nonno R, Bari MA, Cardone F, Vaccari G, Fazzi P, Dell’omo G, Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006;2:e12.. DOIPubMedGoogle Scholar
- Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry. 2005;76:906–19. DOIPubMedGoogle Scholar
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–6. DOIPubMedGoogle Scholar
- Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B, Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc Natl Acad Sci U S A. 2000;97:5574–9. DOIPubMedGoogle Scholar
- Castilla J, Gutierrez-Adan A, Brun A, Pintado B, Salguero FJ, Parra B, Transgenic mice expressing bovine PrP with a four extra repeat octapeptide insert mutation show a spontaneous, non-transmissible, neurodegenerative disease and an expedited course of BSE infection. FEBS Lett. 2005;579:6237–46. DOIPubMedGoogle Scholar
- Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol. 2003;77:7611–22. DOIPubMedGoogle Scholar
- Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science. 1990;250:1587–90. DOIPubMedGoogle Scholar
- Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron. 2009;63:438–50. DOIPubMedGoogle Scholar
- Sigurdson CJ, Nilsson KP, Hornemann S, Heikenwalder M, Manco G, Schwarz P, De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci U S A. 2009;106:304–9. DOIPubMedGoogle Scholar
- Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Linkage of a prion protein missense variant to Gerstmann–Sträussler syndrome. Nature. 1989;338:342–5. DOIPubMedGoogle Scholar
- Ikeda SI, Yanagisawa N, Glenner GG, Allsop D. Gerstmann–Sträussler–Scheinker disease showing protein amyloid deposits in the peripheral regions of PrP immunoreactive amyloid plaques. Neurodegeneration. 1992;1:281–8.
- Castilla J, Gutierrez Adan A, Brun A, Pintado B, Ramirez MA, Parra B, Early detection of PrP(res) in BSE-infected bovine PrP transgenic mice. Arch Virol. 2003;148:677–91. DOIPubMedGoogle Scholar
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–63. DOIPubMedGoogle Scholar
- Castilla J, Gutierrez-Adan A, Brun A, Pintado B, Parra B, Ramirez MA, Different behavior toward bovine spongiform encephalopathy infection of bovine prion protein transgenic mice with one extra repeat octapeptide insert mutation. J Neurosci. 2004;24:2156–64. DOIPubMedGoogle Scholar
- Sisó S, Puig B, Varea R, Vidal E, Acin C, Prinz M, Abnormal synaptic protein expression and cell death in murine scrapie. Acta Neuropathol. 2002;103:615–26. DOIPubMedGoogle Scholar
- Padilla D, Béringue V, Espinosa JC, Andreoletti O, Jaumain E, Reine F, Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog. 2011;7:e1001319. DOIPubMedGoogle Scholar
- Féraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C, Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280:11247–58. DOIPubMedGoogle Scholar
- Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:151–7. DOIPubMedGoogle Scholar
- Andréoletti O, Lacroux C, Chabert A, Monnereau L, Tabouret G, Lantier F, PrP(Sc) accumulation in placentas of ewes exposed to natural scrapie: influence of foetal PrP genotype and effect on ewe-to-lamb transmission. J Gen Virol. 2002;83:2607–16 .PubMedGoogle Scholar
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78:301–11. DOIPubMedGoogle Scholar
- Andréoletti O, Simon S, Lacroux C, Morel N, Tabouret G, Chabert A, PrP(Sc) accumulation in myocytes from sheep incubating natural scrapie. Nat Med. 2004;10:591–3. DOIPubMedGoogle Scholar
- Brun A, Castilla J, Ramirez MA, Prager K, Parra B, Salguero FJ, Proteinase K enhanced immunoreactivity of the prion protein-specific monoclonal antibody 2A11. Neurosci Res. 2004;48:75–83. DOIPubMedGoogle Scholar
- Torres JM, Andreoletti O, Lacroux C, Prieto I, Lorenzo P, Larska M, Classical bovine spongiform encephalopathy by transmission of H-type prion in homologous prion protein context. Emerg Infect Dis. 2011;17:1636–44. DOIPubMedGoogle Scholar
- Espinosa JC, Andreoletti O, Castilla J, Herva ME, Morales M, Alamillo E, Sheep-passaged bovine spongiform encephalopathy agent exhibits altered pathobiological properties in bovine-PrP transgenic mice. J Virol. 2007;81:835–43. DOIPubMedGoogle Scholar
- Bruce ME, McConnell I, Fraser H, Dickinson AG. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol. 1991;72:595–603. DOIPubMedGoogle Scholar
- Priola SA, Chesebro B. Abnormal properties of prion protein with insertional mutations in different cell types. J Biol Chem. 1998;273:11980–5. DOIPubMedGoogle Scholar
- Hsiao KK, Groth D, Scott M, Yang SL, Serban H, Rapp D, Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc Natl Acad Sci U S A. 1994;91:9126–30. DOIPubMedGoogle Scholar
- Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev. 1996;10:1736–50. DOIPubMedGoogle Scholar
- Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 1999;18:6855–64. DOIPubMedGoogle Scholar
- Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M, Houghton R, Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol. 2009;90:546–58. DOIPubMedGoogle Scholar
- Nicholson EM, Brunelle BW, Richt JA, Kehrli ME Jr, Greenlee JJ. Identification of a heritable polymorphism in bovine PRNP associated with genetic transmissible spongiform encephalopathy: evidence of heritable BSE. PLoS ONE. 2008;3:e2912. DOIPubMedGoogle Scholar
- Stöhr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen HO, Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A. 2011;108:21223–8 . DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 19, Number 12—December 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Juan-María Torres, Centro de Investigación en Sanidad Animal, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, 28130 Valdeolmos, Madrid, SpainJuan-María Torres, Centro de Investigación en Sanidad Animal, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, 28130 Valdeolmos, Madrid, Spain
Top