Volume 28, Number 3—March 2022
Research
Genomic and Phenotypic Insights for Toxigenic Clinical Vibrio cholerae O141
Cite This Article
Citation for Media
Abstract
Vibrio cholerae remains a major public health threat worldwide, causing millions of cholera cases each year. Although much is known about the evolution and pathogenicity of the O1/O139 serogroups of V. cholerae, information is lacking on the molecular epidemiology of non‒O1/O139 strains isolated from patients who have diarrheal illnesses. We performed whole-genome sequence analysis and in vivo infections to investigate characteristics of V. cholerae O141 isolated from sporadic diarrheal cases in 4 countries. The strains formed a distinct phylogenetic clade distinguishable from other serogroups and a unique multilocus sequence type 42, but interstrain variation suggests that O141 isolates are not clonal. These isolates encode virulence factors including cholera toxin and the toxin-coregulated pilus, as well as a type 3 secretion system. They had widely variable capacities for intestinal colonization in the infant mouse model. We propose that O141 isolates comprise a distinct clade of V. cholerae non‒O1/O139, and their continued surveillance is warranted.
There are an estimated 3–4 million cases of cholera globally each year, driving marked interest in understanding the genomic diversity and evolution of the causative pathogen (1,2). Of the >200 known Vibrio cholerae serogroups distinguished by unique O-antigen structures, only O1 and O139 have been recognized as being capable of causing sustained epidemics. The O139 serogroup, which caused large epidemics on the Indian subcontinent during 1992–1994, arose from V. cholerae O1 by exchange of the O139 gene cluster encoding O-antigen biosynthesis for the O1 cluster (3). The 2 biotypes of V. cholerae serogroup O1 have been the causes of the previous 6 (classical) and ongoing seventh cholera pandemic (El Tor) (4,5). Decades of study of V. cholerae O1 have showed that cholera pathogenesis is largely driven by the activity of the secreted cholera toxin (Ctx), a potent AB5 toxin that targets intestinal epithelial cells and causes secretory diarrhea in infected hosts. V. cholerae intestinal colonization depends on the toxin-coregulated pilus (Tcp), which is coordinately expressed with Ctx (6).
Compared with information available on V. cholerae O1, relatively little knowledge is available on the pathogenesis and genomic diversity of V. cholerae isolates from other serogroups, such as O37, O75, and O141 (collectively termed non‒O1/O139). These serogroups have been isolated from patients who had diarrheal illness, as well as from aquatic environmental sources (7–10). In the United States, for instance, toxigenic V. cholerae O141 has occasionally been associated with diarrhea and bloodstream infections (11,12). Although non‒O1/O139 strains can encode Ctx and Tcp, they may be underreported as a cause of diarrheal illness because routine laboratory testing in cholera-endemic settings only includes testing for O1 and O139 serogroups (13). Surveillance of non‒O1/O139 serogroups in the United States over the past 30 years has reported diarrheal illness associated with V. cholerae O75 and O141 infection from consumption of seafood or exposure to water in lakes and rivers (8,9).
Previous studies showed that V. cholerae O141 isolates can encode Ctx and Tcp (10,14). In this study, we investigated the genomics and in vivo colonization ability of V. cholerae O141 strains isolated from diarrheal cases from 4 different countries during 1984‒1994. The strains were isolated from sporadic cases of diarrhea without any documented epidemiologic association.
Strain Collection, DNA Extraction, and Whole-Genome Sequencing
We obtained V. cholerae O141 isolates sequenced in the present study from a strain collection initially reported by Dalsgaard et al. (10,15). The strains were isolated from sporadic cases of diarrhea, which did not appear to be epidemiologically related. Information about whether stool samples were cultured for major enteric pathogens other than V. cholerae was not available for the strains studied.
We obtained strains from the Center for Disease Control and Prevention (Atlanta, GA, USA) and the Japanese National Institute of Infectious Diseases (Tokyo, Japan). We stored strains in 10% glycerol at −80°C, and revived them by streaking onto blood agar plates. We extracted genomic DNA from overnight liquid cultures of the isolates by using the Maxwell RSC Cultured Cells DNA kit following the manufacturer’s protocol and the automated Maxwell RSC Machine (both from Promega, https://www.promega.com). We sequenced genomic DNA samples by using the MiSeq System (Illumina, https://www.illumina.com) as described (16). The coverage of the sequenced genomes ranged from 50× to 75× (Table). We submitted the sequence reads to the European Nucleotide Archive (accession no. PRJEB42289).
Read Processing and Genome Assembly
We trimmed raw sequence reads by using with bbduk2 (17) (from BBmap version 6.49) and a cutoff score of 20. We evaluated read quality by using FastQC version 0.11.5 (https://guix.gnu.org) before and after trimming. We assembled trimmed reads by using Spades version 3.13.0 (18), error correction, a coverage cutoff of 2, and kmer sizes 21, 33, 55, 77, 99 and 127. We discarded contigs <200 bases and assessed the quality of the de novo assembled contigs by using Quast version 4.5 (19). We then analyzed the assembled genomes for species identification and V. cholerae–specific genome annotation (biotype, serogroup, and Vibrio pathogenicity island conservation) by using the CholeraeFinder tool (https://cge.cbs.dtu.dk/services/CholeraeFinder). We identified resistance genes by using ResFinder (20) and plasmid replicons by using PlasmidFinder (21).
Phylogenetic Analysis
We used the generated V. cholerae O141 genomes for phylogenetic analysis with publicly available genomes representing the other Ctx-positive V. cholerae serogroups. Representative clinical nontoxigenic and non‒O1/O139 genomes from strains isolated in Germany were also included in the analysis (22). We analyzed 23 additional ctxA-positive V. cholerae and 7 ctxA-negative non‒O1/0139 reference genomes and compared them with the 8 genomes we had (total = 38). These genomes included the only whole genomes sequences of V. cholerae O141 available before this study (strain V51 and 234–93), all publicly available genomes of V. cholerae O75 and O37 (all ctxA+ non‒O1/O139 serogroups), the representative O139 strain MO10, and a variety of historical and contemporary O1 strains with differing ctxB alleles, which were selected to capture the genomic variation of pandemic V. cholerae O1. These historical and contemporary O1 strains included strains O395 (classical, ctxB1), N16961 (El Tor, ctxB3), CTMA1422 (El Tor variant, ctxB1), L254 (El Tor variant, ctxB1) and ZB6 (El Tor variant, ctxB7). We provide details and accession numbers of these genomes, including the nontoxigenic non‒O1/O139 strains (Appendix Table 1).
We called single-nucleotide variants by using Snippy version 4.6.0 (https://github.com/tseemann/snippy) under the following parameters: mapping quality of 60, a minimum base quality of 13, a minimum read coverage of 4, and a 75% concordance at a locus. We aligned core genome single-nucleotide variants by using Snippy version 4.1.0 for phylogeny inference. We detected masked putative recombinogenic regions by using Gubbins version 2.4.1 (23). We built a maximum-likelihood phylogenetic tree by using RAxML version/8.2.12 and the generalized time-reversible model with 100 bootstraps (24). We rooted the final tree on the V51 genome and visualized it with iTOL version 3 (25). We provide pairwise single-nucleotide polymorphism (SNP) data for the 38 strains (Appendix Table 2). The alignment length from all analyzed genomes was 3,464,958 and represented 82.3% of the reference V. cholerae strain V51 used.
Comparative Genomics
We annotated all genomes used for phylogenetic analysis by using Prokka version 1.14.5 (26), and used resulting general feature format 3 files as inputs to the Roary version 3.7.0 (27) pangenome analysis tool. We then used the binary presence/absence data of the accessory genome produced in Roary to calculate associations between all genes in the accessory genome and serogroups by using Scoary version 1.6.11 (28). We depicted a heatmap of the genes present or absent in the core genome, along with the accessory genome, in phandango (29) to enable the identification and extraction of the unique coding sequence (CDS) blocks observed for the O141 serogroup by applying the query_pan_genome function of Roary. After a BLAST Atlas analysis from the GView server (https://server.gview.ca), we mapped the multi-FASTA files of the O141-specific CDS block to the reference V51 to localize the block in the genome.
To understand how O141-specific CDS could play a role in intestinal colonization, we analyzed the extracted multi-FASTA file by using the VRprofile pipeline (30), which detects virulence and colonization determinants within bacterial genomes. We customized this analysis to focus on the gene clusters encoding Tcp, T3SS2 a Vibrio type III secretion system that is found in clinical V. parahaemolyticus isolates and in some V. cholerae non‒O1/O139, and other accessory colonization factors known to promote V. cholerae intestinal colonization (31–34). In addition, we individually investigated genes/open reading frames located in these clusters by using local blastn and blastp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) searches against our query genomes with intentionally low 60% query cover and 30% identity thresholds to avoid false-negative gene, absence outcomes that might be caused by recombination.
Infant Mouse Intestinal Colonization Assay
We orally inoculated 5-day-old, infant CD-1 mice (Charles River Laboratories https://www.criver.com) with V. cholerae as described (35). We used frozen stocks of each strain to inoculate lysogeny broth that did not contain antimicrobial dugs and incubated the broth overnight with shaking at 250 rpm at 37°C. We diluted cultures 1:1,000 in lysogeny broth and mixed the cultures with 4 μL/mL of green food coloring to track the inoculum. We removed pups from their dams 1 hour preinoculation and orally inoculated them with 50 μL of diluted culture (≈2–4 ×105 CFU/pup). We combined and randomly assigned pups from multiple litters to inoculation groups to reduce the effect of litter effects on V. cholerae colonization. We housed inoculated pups in a warmed box with nest material for 20 hours in the dark apart from their dams, at which point they were euthanized with isoflurane inhalation followed by decapitation. We dissected and mechanically homogenized small intestines by using a Tissue Tearor (BioSpec, https://biospec.com), followed by serial dilution and bead plating onto thiosulfate-citrate-bile salt (TCBS) agar plates that did not contain antimicrobial drugs. We incubated plates at 37°C overnight for counting. No non-Vibrio (non-yellow) colonies were detected on the TCBS agar plates. Animal work in this study was approved by the Brigham and Women’s Hospital Institutional Animal Care and Use Committee under Protocol #2016N000416.
Genomic Characterization and Phylogenetic Analysis
To investigate the genomic diversity of clinical isolates of V. cholerae O141, we sequenced and annotated the genomes of 8 serotype-confirmed O141 strains collected from stool samples of gastroenteritis patients in the United States, Spain, Taiwan, and India over a 10-year period during 1984–1994 (10) (Table). These strains had been characterized by using ribotyping, PCRs for ctxA and tcpA, and antimicrobial drug susceptibility testing, but little was known about their genomic characteristics (10,15). All 8 isolates had gene sequences in the O-antigen lipopolysaccharide region and gene rearrangements between gmhD and wbfY, consistent with known O141-specific lipopolysaccharide changes (Appendix Table 3) (9,36). Sequence typing also placed all 8 isolates in the same multilocus sequence type (MLST), MLST42, as the known O141 isolate V51 (Table). On the basis of concordance in the 7-gene MLST profile, these observations suggest that ST42 might be specific to the serogroup O14, and could serve as a serogroup-specific marker for genomic studies because no other V. cholerae serogroups have been associated with this MSLT (2,16,37).
Figure 1
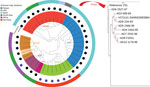
Figure 1. Maximum-likelihood phylogenetic tree for Vibrio cholerae O141 in a global context for 38 isolates from Ctx-positive V. choleraeand Ctx-negative serogroups. Numbers along branches are bootstrap values....
The core genome MLST, which is based on the entire core genome rather than the 7 housekeeping genes used for conventional MLST, was cgST-479 for all except the strains AD8 (cgST-246) and V51 (cgST-248). This variation was further reflected in the whole-genome phylogenetic analysis, in which O141 strains, although distinct from other serogroups, were not internally clonal, differing in up to 261 SNPs (Figure 1).
Despite their diverse sites and years of isolation, all 8 O141 strains encoded a CTX prophage similar to the classical CTX prophage with the ctxB1 allele and the classical rstR as indicated (38). The presence of the classical CTX prophage in all 8 strains suggests that the presence of this sequence along with the alleles that constitute MLST42 might be characteristic of serogroup O141. In addition to ctxAB, the genes encoding the signature virulence factor of V. cholerae, these strains also encoded cholix toxin, an accessory toxin that is found in V. cholerae non‒O1/O139 (39). Although these strains harbored a classical CTX prophage, they all also contained an El Tor type tcpA, which encodes the major subunit of the Tcp pilus, the CTXϕ receptor, and a critical determinant of intestinal colonization V. cholerae O1 (40). Although most strains also contained genes in the tcp operon needed for Tcp biogenesis (encoded in the Vibrio pathogenicity island VPI-I), they generally lacked an intact tcpJ, which encodes a prepilin peptidase required for processing of TcpA (41). All sequenced strains also appeared to encode a type III secretion system (T3SS) known as T3SS2, that is a critical colonization and virulence determinant of V. parahaemolyticus and is also found in V51 (34,42). The co-occurrence of the TCP and T3SS2 pathogenicity islands in V. cholerae O141 strains suggests that V. cholerae O141 might rely on diverse mechanisms for pathogenicity, potentially deploying these distinct virulence mechanisms in different hosts.
The O141 strains did not contain detectable antimicrobial resistance genes, supporting prior phenotypic antimicrobial drug susceptibility findings in which all strains were susceptible to a panel of 12 antimicrobial drugs, except for colistin (to which all non‒O1 V. cholerae naturally show resistance) (10). In addition, none of the analyzed V. cholerae O141 genomes contained plasmid replicons, consistent with the absence of plasmids, as shown by previous plasmid extraction analysis of these isolates (10).
Despite the observed homogeneity in MLST profile and conservation of major virulence genes in V. cholerae O141 strains, there were substantial variations in the O141 genomes (up to 261 SNPs), regardless of country of origin (Figure 1; Appendix Table 2), most of which occurred in noncoding regions. This finding suggests that the strains are epidemiologically unrelated, consistent with the idea that infections caused by V. cholerae O141 are sporadic. All the O141 serogroup strains, including V51, formed a separate clade distinguishable from the other serogroups, all strains from serogroup O75 also grouped into a distinct clade (Figure 1). The observed genetic variations between the serogroups indicates that V. cholerae O141 and O75 are not phylogenetically related, contrary to a previous proposal (8). The phylogeny also suggests that serogroup O37 is closely related to the classical O1 strain O395. As expected, serogroup O139 represented by the reference strain MO10 was localized to the O1 El Tor subclade, consistent with the idea that this serogroup arose from an O1 El Tor seventh pandemic strain (3,21,37). Moreover, the nontoxigenic non‒O1/O139 clinical strains formed a separate clade on the phylogenetic tree that is unrelated to the other known toxigenic, as well as nontoxigenic serogroups.
Intestinal Colonization of Infant Mice by V. cholerae Serogroup O141
Figure 2
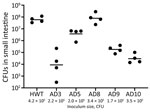
Figure 2. Intestinal colonization of 5-day-old infant mice by Vibrio choleraeO141. Pups were orally inoculated with the indicated amount of the indicated strain, and CFUs in the small intestine were...
The presence of canonical pandemic V. cholerae colonization factors such as Tcp in their genomes led us to hypothesize that O141 strains, like their pandemic O1 counterparts, might colonize the small intestine. To test this idea, we used the well-characterized infant mouse model of V. cholerae small intestinal colonization. Infant mice orally inoculated with 2–4 × 105 CFU of selected O141 strains that grew well on TCBS agar plates (AD3, AD5, AD8, AD9, and AD10) showed marked variation in their colonization capacity (Figure 2). In comparison to a V. cholerae O1 isolate from the recent cholera epidemic in Haiti, which robustly colonizes the small intestine (43), strains AD8 and AD5 had similar numbers of CFU recovered in intestinal homogenates as the strain from Haiti (Figure 2). In contrast, the other 3 strains had from ≈1,000-fold (AD9) to ≈10,000-fold (AD3 and AD10) lower numbers of recoverable bacteria, indicating that although they can all colonize the small intestine, there are considerable strain-specific differences in the capacities of these O141 isolates to colonize the mammalian small intestine.
Figure 3

Figure 3. Gene presence/absence map of the pangenome of 31 isolates from cholera toxin‒positive Vibrio choleraeserogroups. The red rectangle in the accessory genome indicates the conserved unique coding sequences that...
Figure 4

Figure 4. Targeted analysis of the accessory genome of in vivo‒tested Vibrio choleraeO141 isolates. A) Genes encoding colonization factors. For each block of colonization factor, the absent genes are represented...
Differential genomic conservation of virulence or colonization determinants could underlie the variable colonization phenotypes. To evaluate strain-level conservation of accessory genetic features, we next performed pangenome analysis of genomes from only the toxigenic strains used in the phylogenetic analysis. This analysis identified an accessory genome made of shell and cloud genes of 2,598 coding sequences (CDS) in a total pangenome size of 5,627 CDS (Figure 3; Appendix Table 4). A targeted analysis of the accessory genome showed strain-specific gene absences in the in vivo‒tested O141 strains (Figure 4). For example, AD3, which had the lowest intestinal colonization among the strains tested, lacked toxT, the master transcription activator of V. cholerae virulence genes (44) (Figure 4). The accessory genomes of AD3, AD9, and AD10, which did not colonize as well as the robustly colonizing strains AD5 and AD8, all lacked T3SS2 genes vcrS2 and vopB2 (Figure 4, panel A). AD3 also lacked the known T3SS effectors vopF and sseJ (Figure 4, panel A). All analyzed strains, including V51, contained protein sequences corresponding to VopV and VopZ, 2 T3SS2-associated genes known to be critical for intestinal colonization by V. parahaemolyticus (34,42).
Our findings show that V. cholerae O141 clinical isolates form a genetically distinct clade that is distinguishable from pandemic and nonpandemic V. cholerae serogroups. The observation that all tested isolates encoded known virulence factors and were capable of colonizing the infant mouse intestine, albeit in a highly variable manner, supports the idea that V. cholerae O141 could be an underestimated source of cholera-like diarrhea. Currently, O141 cases would be grouped under the umbrella of non‒O1/O139 cases because of a lack of widely available serogroup-specific antiserum for O141. Nevertheless, from this study, the ST42 that appears to be specific/unique to the serogroup O141 might be used for diagnostic purposes as an alternative to O141 antiserum, which is not widely available.
Our findings show that some O141 strains are capable of robust colonization. These strains encode at least 2 potential mechanisms, Tcp and T3SS2, that could enable intestinal colonization. Variable colonization among O141 strains could be explained by differential conservation of T3SS components/effectors or other colonization factors. Deciphering the colonization requirements of different O141 isolates will be a useful endeavor.
The factors that have limited V. cholerae O141 from causing sustained cholera epidemics remain to be elucidated. It is possible that V. cholerae O141 is not as well adapted as V. cholerae O1 to the aquatic environment, which is thought to be a key feature of the lifecycle of V. cholerae. Although we did not assess the aquatic fitness of the O141 serogroup, V. cholerae O141 has been detected in environmental reservoirs, such as oysters, clams, and freshwater in lakes and rivers in the United States, suggesting an environmental defect is unlikely to fully explain the low frequency of these strains in the clinic (8,9). These discrepancies call for further genomic and experimental studies on environmental, as well as additional clinical V. cholerae O141 isolates. Additional techniques, such as multilocus sequence typing, could overcome challenges related to the identification of V. cholerae non‒O1/O139 serogroups.
Overall, V. cholerae O141 strains constitute a distinct phylogenetic clade that includes shared and unique genomic elements. In addition, we found that V. cholerae O141 clinical isolates showed marked variation in intestinal colonization capacity in the infant mouse model. These findings shed light on a little-known V. cholerae serogroup associated with diarrheal illness.
Dr. Hounmanou is a postdoctoral fellow at the Department of Veterinary and Animal Sciences, University of Copenhagen, Frederiksberg, Denmark. His primary research interests are One Health; microbial genomics; human bacterial pathogens that arise from animals and the aquatic environment; and the ecology and routes of transmission between animals, humans, and waterbodies, with a specific focus on V. cholerae.
Acknowledgment
This study was supported by the University of Copenhagen. M.K.W. was supported by the National Institutes of Health (grant AI-042347) and the Howard Hughes Medical Institute.
References
- Ali M, Nelson AR, Lopez AL, Sack DA. Updated global burden of cholera in endemic countries. PLoS Negl Trop Dis. 2015;9:
e0003832 . DOIPubMedGoogle Scholar - Weill F-X, Domman D, Njamkepo E, Almesbahi AA, Naji M, Nasher SS, et al. Genomic insights into the 2016-2017 cholera epidemic in Yemen. Nature. 2019;565:230–3. DOIPubMedGoogle Scholar
- Faruque SM, Sack DA, Sack RB, Colwell RR, Takeda Y, Nair GB. Emergence and evolution of Vibrio cholerae O139. Proc Natl Acad Sci U S A. 2003;100:1304–9. DOIPubMedGoogle Scholar
- Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–5. DOIPubMedGoogle Scholar
- Grad YH, Waldor MK. Deciphering the origins and tracking the evolution of cholera epidemics with whole-genome-based molecular epidemiology. MBio. 2013;4:e00670–13. DOIPubMedGoogle Scholar
- Faruque SM, Albert MJ, Mekalanos JJ. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol Mol Biol Rev. 1998;62:1301–14. DOIPubMedGoogle Scholar
- Fang L, Ginn AM, Harper J, Kane AS, Wright AC. Survey and genetic characterization of Vibrio cholerae in Apalachicola Bay, Florida (2012-2014). J Appl Microbiol. 2019;126:1265–77. DOIPubMedGoogle Scholar
- Crowe SJ, Newton AE, Gould LH, Parsons MB, Stroika S, Bopp CA, et al. Vibriosis, not cholera: toxigenic Vibrio cholerae non-O1, non-O139 infections in the United States, 1984-2014. Epidemiol Infect. 2016;144:3335–41. DOIPubMedGoogle Scholar
- Haley BJ, Choi SY, Grim CJ, Onifade TJ, Cinar HN, Tall BD, et al. Genomic and phenotypic characterization of Vibrio cholerae non-O1 isolates from a US Gulf Coast cholera outbreak. PLoS One. 2014;9:
e86264 . DOIPubMedGoogle Scholar - Dalsgaard A, Serichantalergs O, Forslund A, Lin W, Mekalanos J, Mintz E, et al. Clinical and environmental isolates of Vibrio cholerae serogroup O141 carry the CTX phage and the genes encoding the toxin-coregulated pili. J Clin Microbiol. 2001;39:4086–92. DOIPubMedGoogle Scholar
- Crump JA, Bopp CA, Greene KD, Kubota KA, Middendorf RL, Wells JG, et al. Toxigenic Vibrio cholerae serogroup O141-associated cholera-like diarrhea and bloodstream infection in the United States. J Infect Dis. 2003;187:866–8. DOIPubMedGoogle Scholar
- Loeck BKD, Roberts A, Craney AR, King S, Im MS, Safranek TJ, et al. Notes from the field: toxigenic Vibrio cholerae O141 in a traveler to Florida—Nebraska, 2017. MMWR Morb Mortal Wkly Rep. 2018;67:838–9. DOIPubMedGoogle Scholar
- Chen M, Guo D, Wong HC, Zhang X, Liu F, Chen H, et al. Development of O-serogroup specific PCR assay for detection and identification of Vibrio parahaemolyticus. Int J Food Microbiol. 2012;159:122–9. DOIPubMedGoogle Scholar
- Udden SM, Zahid MS, Biswas K, Ahmad QS, Cravioto A, Nair GB, et al. Acquisition of classical CTX prophage from Vibrio cholerae O141 by El Tor strains aided by lytic phages and chitin-induced competence. Proc Natl Acad Sci U S A. 2008;105:11951–6. DOIPubMedGoogle Scholar
- Dalsgaard A, Forslund A, Mortensen HF, Shimada T. Ribotypes of clinical Vibrio cholerae non-O1 non-O139 strains in relation to O-serotypes. Epidemiol Infect. 1998;121:535–45. DOIPubMedGoogle Scholar
- Hounmanou YMG, Leekitcharoenphon P, Kudirkiene E, Mdegela RH, Hendriksen RS, Olsen JE, et al. Genomic insights into Vibrio cholerae O1 responsible for cholera epidemics in Tanzania between 1993 and 2017. PLoS Negl Trop Dis. 2019;13:
e0007934 . DOIPubMedGoogle Scholar - Bushnell B, Rood J, Singer E. BBMerge - Accurate paired shotgun read merging via overlap. PLoS One. 2017;12:
e0185056 . DOIPubMedGoogle Scholar - Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. DOIPubMedGoogle Scholar
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–5. DOIPubMedGoogle Scholar
- Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. 2020;75:3491–500. DOIPubMedGoogle Scholar
- Siriphap A, Leekitcharoenphon P, Kaas RS, Theethakaew C, Aarestrup FM, Sutheinkul O, et al. Characterization and genetic variation of Vibrio cholerae isolated from clinical and environmental sources in Thailand. PLoS One. 2017;12:
e0169324 . DOIPubMedGoogle Scholar - Schwartz K, Hammerl JA, Göllner C, Strauch E. Environmental and clinical strains of Vibrio cholerae non-O1, non-O139 from Germany possess similar virulence gene profiles. Front Microbiol. 2019;10:733. DOIPubMedGoogle Scholar
- Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:e15–15. DOIPubMedGoogle Scholar
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. DOIPubMedGoogle Scholar
- Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):
W242-5 . DOIPubMedGoogle Scholar - Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. DOIPubMedGoogle Scholar
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3. DOIPubMedGoogle Scholar
- Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016;17:238. DOIPubMedGoogle Scholar
- Hadfield J, Croucher NJ, Goater RJ, Abudahab K, Aanensen DM, Harris SR. Phandango: an interactive viewer for bacterial population genomics. Bioinformatics. 2018;34:292–3. DOIPubMedGoogle Scholar
- Li J, Tai C, Deng Z, Zhong W, He Y, Ou H-Y. VRprofile: gene-cluster-detection-based profiling of virulence and antibiotic resistance traits encoded within genome sequences of pathogenic bacteria. Brief Bioinform. 2018;19:566–74.PubMedGoogle Scholar
- Shakhnovich EA, Sturtevant D, Mekalanos JJ. Molecular mechanisms of virstatin resistance by non-O1/non-O139 strains of Vibrio cholerae. Mol Microbiol. 2007;66:1331–41. DOIPubMedGoogle Scholar
- Shin OS, Tam VC, Suzuki M, Ritchie JM, Bronson RT, Waldor MK, et al. Type III secretion is essential for the rapidly fatal diarrheal disease caused by non-O1, non-O139 Vibrio cholerae. MBio. 2011;2:e00106–11. DOIPubMedGoogle Scholar
- Zhou X, Gewurz BE, Ritchie JM, Takasaki K, Greenfeld H, Kieff E, et al. A Vibrio parahaemolyticus T3SS effector mediates pathogenesis by independently enabling intestinal colonization and inhibiting TAK1 activation. Cell Rep. 2013;3:1690–702. DOIPubMedGoogle Scholar
- Hiyoshi H, Kodama T, Saito K, Gotoh K, Matsuda S, Akeda Y, et al. VopV, an F-actin-binding type III secretion effector, is required for Vibrio parahaemolyticus-induced enterotoxicity. Cell Host Microbe. 2011;10:401–9. DOIPubMedGoogle Scholar
- Fleurie A, Zoued A, Alvarez L, Hines KM, Cava F, Xu L, et al. A Vibrio cholerae BolA-like protein is required for proper cell shape and cell envelope integrity. MBio. 2019;10:10. DOIPubMedGoogle Scholar
- Aydanian A, Tang L, Morris JG, Johnson JA, Stine OC. Genetic diversity of O-antigen biosynthesis regions in Vibrio cholerae. Appl Environ Microbiol. 2011;77:2247–53. DOIPubMedGoogle Scholar
- Mutreja A, Dougan G. Molecular epidemiology and intercontinental spread of cholera. Vaccine. 2020;38(Suppl 1):A46–51. DOIPubMedGoogle Scholar
- Davis BM, Kimsey HH, Kane AV, Waldor MK. A satellite phage-encoded antirepressor induces repressor aggregation and cholera toxin gene transfer. EMBO J. 2002;21:4240–9. DOIPubMedGoogle Scholar
- Awasthi SP, Asakura M, Chowdhury N, Neogi SB, Hinenoya A, Golbar HM, et al. Novel cholix toxin variants, ADP-ribosylating toxins in Vibrio cholerae non-O1/non-O139 strains, and their pathogenicity. Infect Immun. 2013;81:531–41. DOIPubMedGoogle Scholar
- Clemens JD, Nair GB, Ahmed T, Qadri F, Holmgren J. Cholera. Lancet. 2017;390:1539–49. DOIPubMedGoogle Scholar
- Kaufman MR, Seyer JM, Taylor RK. Processing of TCP pilin by TcpJ typifies a common step intrinsic to a newly recognized pathway of extracellular protein secretion by gram-negative bacteria. Genes Dev. 1991;5:1834–46. DOIPubMedGoogle Scholar
- Zhou X, Massol RH, Nakamura F, Chen X, Gewurz BE, Davis BM, et al. Remodeling of the intestinal brush border underlies adhesion and virulence of an enteric pathogen. MBio. 2014;5:5. DOIPubMedGoogle Scholar
- Sit B, Zhang T, Fakoya B, Akter A, Biswas R, Ryan ET, et al. Oral immunization with a probiotic cholera vaccine induces broad protective immunity against Vibrio cholerae colonization and disease in mice. PLoS Negl Trop Dis. 2019;13:
e0007417 . DOIPubMedGoogle Scholar - Matson JS, Withey JH, DiRita VJ. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect Immun. 2007;75:5542–9. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: February 10, 2022
Table of Contents – Volume 28, Number 3—March 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Yaovi M.G. Hounmanou, Department of Veterinary and Animal Sciences, University of Copenhagen, 1870 Frederiksberg, Denmark
Top