Volume 16, Number 7—July 2010
Research
High Diversity and Ancient Common Ancestry of Lymphocytic Choriomeningitis Virus
Figure 1
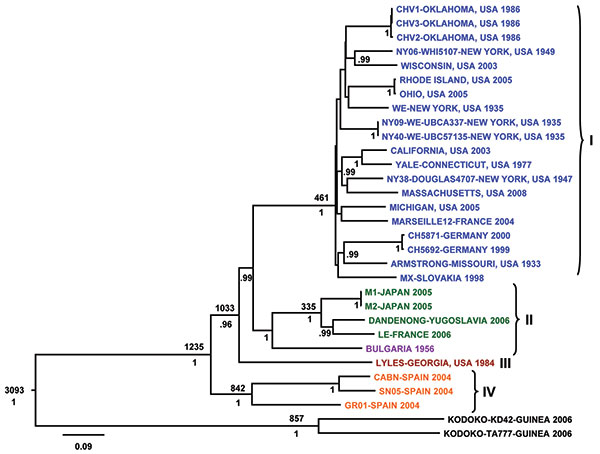
Figure 1. Bayesian coalescent analysis of lymphocytic choriomeningitis virus (LCMV) based on the small (S) gene segment. The maximum clade credibility tree generated from analysis of available LCMV S segment sequences is shown. Branch lengths are proportional to the number of substitutions/site/year. Depicted at the main nodes are the time to most recent common ancestor estimates (TMRCA) based on Bayesian coalescent analysis of the virus sequences and isolation dates without inclusion of the Bulgarian strain for which no reliable isolation date was available. Posterior probabilities are listed below the branches for supported nodes. Scale bar indicates nucleotide substitutions per site.
1These authors contributed equally to this article.
2Current affiliation: University of Texas Medical Branch, Galveston, Texas, USA.