Volume 20, Number 2—February 2014
Research
Novel Paramyxovirus Associated with Severe Acute Febrile Disease, South Sudan and Uganda, 2012
Figure 2
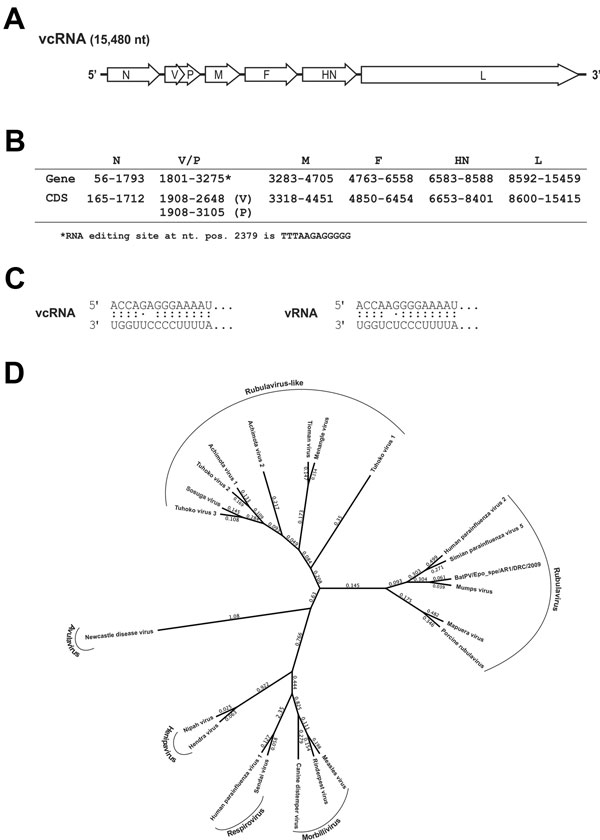
Figure 2. A) Organization of the viral genome of novel paramyxovirus related to rubula-like viruses isolated from fruit bats was determined from the full-length sequenceB) Localization of the predicted viral genes and open reading frames (ORFs)The V/P edition site is predicted from the similarity to Tuhoko virus 3C) Terminal sequences were determined by standard rapid amplification of cDNA ends (RACE) methodsThe complementarity of terminal sequences is shown in vRNA and vcRNA senseD) Amino acid sequences of the nucleocapsid (N) protein of 22 representative paramyxovirus sequences were aligned by using the MUSCLE algorithm (CLC Genomics Workbench version 6.0.1; CLC bio, Cambridge, MA, USA)The phylogenetic analysis was conducted with a Bayesian algorithm (Mr.Bayes, Geneious version 6.1.5, www.geneious.com/)NP sequences were extracted from the complete genomic sequences in GenBank: KF774436 (Sosuga virus [SosV]), GU128082 (Tuhoko virus 3 ), GU128081 (Tuhoko virus 2), GU128080 (Tuhoko virus 1), AF298895 (Tioman virus), NC_007620 (Menangle virus), JX051319 (Achimota virus 1), JX051320 (Achimota virus 2), NC_003443 (human parainfluenza virus type 2), AF052755 (simian parainfluenza virus 5), HQ660095 (bat paramyxovirus Epo_spe/AR1/DRC/2009), NC_002200 (mumps virus), NC_009489 (Mapuera virus), NC_009640 (porcine rubulavirus), NC_001498 (measles virus), NC_006296 (rinderpest virus), NC_001921 (canine distemper virus), NC_001552 (Sendai virus), NC_003461 (human parainfluenza virus type 1), NC_002728 (Nipah virus), NC_001906 (Henra virus), NC_002617 (Newcastle disease virus)vcRNA, viral complementary RNA; N, nucleocapsid protein; V/P, V protein; M, matrix protein; F, fusion protein; HN, hemagglutinin-neuraminidase; L, molecular weight DNA ladder; CDS, coding sequence; nt posnucleotide position; vRNA, viral RNA.