Volume 20, Number 7—July 2014
Letter
Genome Analysis of Mayaro Virus Imported to Germany from French Guiana
Figure
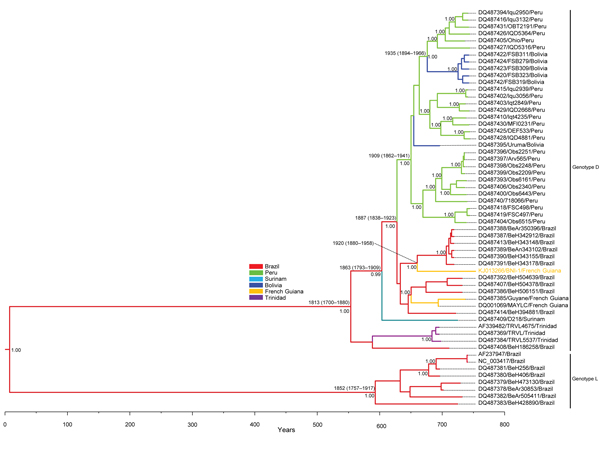
Figure. Bayesian maximum clade credibility tree representing the time-scale phylogeny of Mayaro virus (MAYV) by analysis of 2-kb genomic fragments, including the 3′ segment of the envelope (E)2 protein, the complete E1 protein, and the 3′ non-coding region (NCR)Phylogenetic analysis was performed by using Bayesian Markov chain Monte Carlo (MCMC) tree-sampling method implemented in BEAST (http://beast.bio.ed.ac.uk)The general time reversible model of nucleotide substitution with gamma distributed rate variation among sites and a relaxed (uncorrelated log-normal) molecular clock model were usedTwo independent runs of 5 × 107 generations with a burn in of 5 × 106 generations were performed to estimate the posterior probability distributionConvergence of parameters was confirmed by calculating the effective sample size with Tracer v1.4 (http://tree.bio.ed.ac.uk/software/tracer/) and excluding an initial 10% for each runThe maximum clade credibility tree obtained (tree with the largest product of posterior clade probabilities) was selected from the posterior tree distribution after a 10% burn in by using the TreeAnnotator (http://beast.bio.ed.ac.uk/TreeAnnotator)Taxon information includes GenBank accession number, strain designation, and country of originBranches are colored on the basis of the most probable location state of the descendent nodes (see color codes)The posterior probabilities (clade credibilities ≥90%) and the time to most recent common ancestor of the major branches are shownDate of divergence (95% highest posterior density) of the genotypes, subtypes, and the MAYV strain isolated in this study (in orange) are givenScale bar indicates years before the last sampling time (2013).
1These authors contributed equally to this article.