Chikungunya Virus as Cause of Febrile Illness Outbreak, Chiapas, Mexico, 2014
Tiffany F. Kautz
1, Esteban E. Díaz-González
1, Jesse H. Erasmus, Iliana R. Malo-García, Rose M. Langsjoen, Edward I. Patterson, Dawn I. Auguste, Naomi L. Forrester, Rosa Maria Sanchez-Casas, Mauricio Hernández-Ávila, Celia M. Alpuche-Aranda, Nikos Vasilakis

, and Ildefonso Fernández-Salas
Author affiliations: University of Texas Medical Branch, Galveston, Texas, USA (T.F. Kautz, J.H. Erasmus, R.M. Langsjoen, E.I. Patterson, D.I. Auguste, N.L. Forrester, S.C. Weaver); Universidad Autónoma de Nuevo León, San Nicolás de los Garza, Mexico (E.E. Diaz-Gonzalez); Centro Regional de Investigación en Salud Pública, Tapachula, Mexico (I.R. Malo-García, I. Fernandez-Salas); Universidad Autónoma de Nuevo León, Escobedo, Mexico (R.M. Sanchez-Casas); Instituto Nacional de Salud Pública, Cuernavaca, México (M. Hernández-Ávila, C.M. Alpuche-Aranda)
Main Article
Figure 2
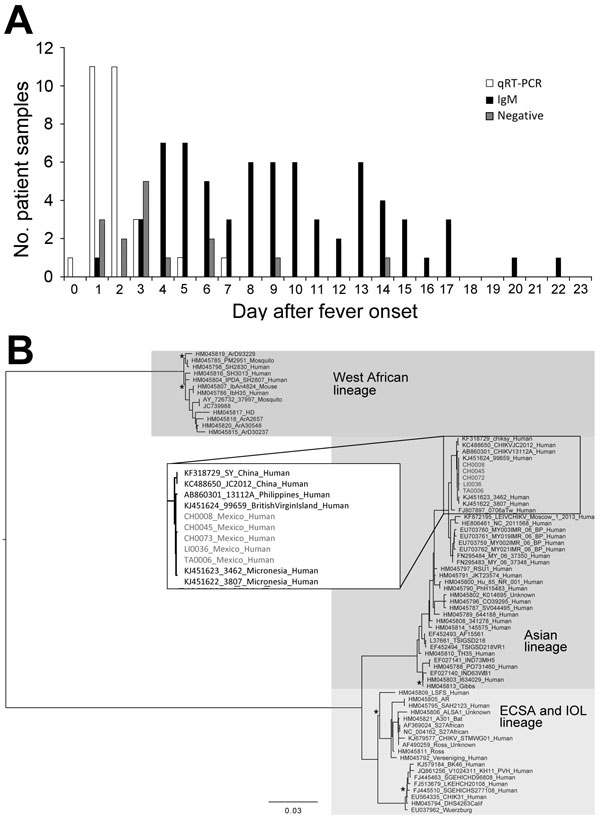
Figure 2. A) Number of serum samples positive for chikungunya virus (CHIKV) by reverse transcription quantitative PCR (qRT-PCR), for CHIKV IgM by ELISA, and negative for CHIKV by both methods, arranged according to day after fever onset. B) Phylogenetic tree generated by Bayesian analysis showing the relationship of the complete genomic sequences of 5 chikungunya virus isolates from Mexico and representative sequences from the GenBank library. All nodes showed posterior probabilities of >0.9, except those indicated with a star. The inset shows the closest relatives of the 5 isolates. ECSA, East/Central/South African, IOL, Indian Ocean lineage. Scale bars indicates nucleotide substitutions per site.
Main Article
Page created: October 19, 2015
Page updated: October 19, 2015
Page reviewed: October 19, 2015
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.