Volume 22, Number 11—November 2016
Letter
Severe Pneumonia Associated with Adenovirus Type 55 Infection, France, 2014
Figure
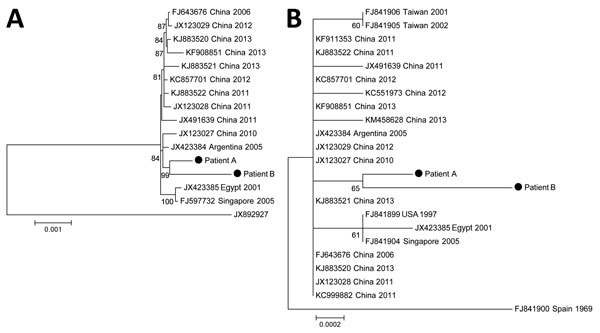
Figure. Phylogeny of 13 complete-genome sequences of human adenovirus type 55 (A) and of 21 sequences of hexon genes (B). The complete genome tree (A) is rooted to a human adenovirus type 14 isolate (GenBank accession no. JX892927). The strains from patients A and B (immunocompetent women with human adenovirus infection) reported in this study are indicated. The phylogenetic tree was calculated by using the maximum-likelihood method in MEGA6 (http://www.megasoftware.net). The best algorithm was chosen by the criterion score of the Bayesian information criteria. The statistical robustness of branches was estimated by 1,000 bootstraps. Only bootstrap values >70% are indicated. The tree is drawn to scale; branch lengths are measured in number of substitutions per site (scale bar). All positions containing gaps and missing data were eliminated. The sequence of the hexon gene from patient B was partially complete (2,821/2,841, 99.3%).