Volume 23, Number 8—August 2017
Dispatch
Global Spread of Norovirus GII.17 Kawasaki 308, 2014–2016
Martin C.W. Chan, Yunwen Hu, Haili Chen, Alexander T. Podkolzin, Ekaterina V. Zaytseva, Jun Komano, Naomi Sakon, Yong Poovorawan, Sompong Vongpunsawad, Thanundorn Thanusuwannasak, Joanne Hewitt, Dawn Croucher, Nikail Collins, Jan Vinjé, Xiaoli L. Pang, Bonita E. Lee, Miranda de Graaf, Janko van Beek, Harry Vennema, Marion P.G. Koopmans, Sandra Niendorf, Mateja Poljsak-Prijatelj, Andrej Steyer, Peter A. White, Jennifer H. Lun, Janet Mans, Tin-Nok Hung, Kirsty Kwok, Kelton Cheung, Nelson Lee, and Paul K.S. Chan
Figure 1
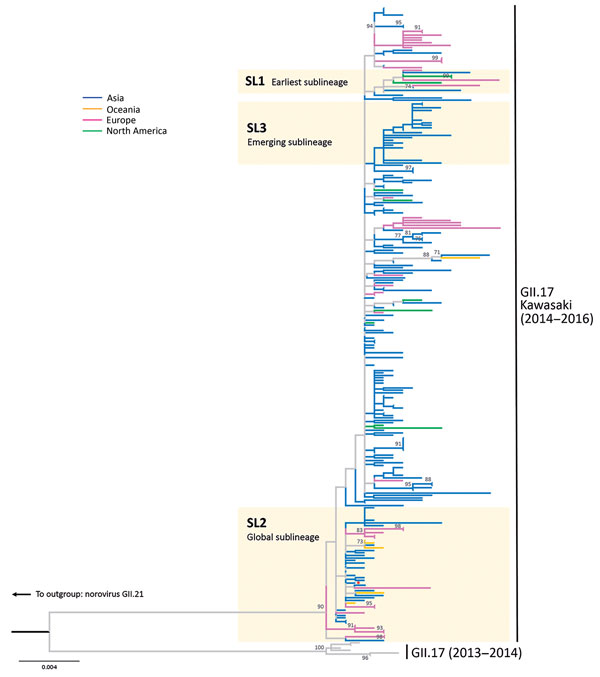
Figure 1. Maximum-likelihood phylogenetic inference of complete viral protein 1 nucleotide sequences of norovirus GII.17 Kawasaki. The tree was constructed using MEGA6 (http://www.megasoftware.net) (Technical Appendix). The red asterisk denotes the reference sequence of GII.17 Kawasaki virus (Hu/GII/JP/2015/GII.P17_GII.17/Kawasaki308; GenBank accession no. LC037415). The tree is rooted to genotype GII.21 (not shown for clarity). Bootstrap values >70 (percentage) are shown at nodes. Sublineages SL1 to SL3 are defined by the topology of haplotype network shown in Figure 2. Branches are colored by the continent of sequence origin. The tree is drawn in scale; scale bar indicates nucleotide substitutions per site.
Page created: July 17, 2017
Page updated: July 17, 2017
Page reviewed: July 17, 2017
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.