Volume 25, Number 1—January 2019
Dispatch
Isolation and Full-Genome Characterization of Nipah Viruses from Bats, Bangladesh
Danielle E. Anderson1, Ariful Islam1, Gary Crameri1, Shawn Todd, Ausraful Islam, Salah U. Khan, Adam Foord, Mohammed Z. Rahman, Ian H. Mendenhall, Stephen P. Luby, Emily S. Gurley, Peter Daszak, Jonathan H. Epstein1 , and Lin-Fa Wang1
, and Lin-Fa Wang1
Figure 2
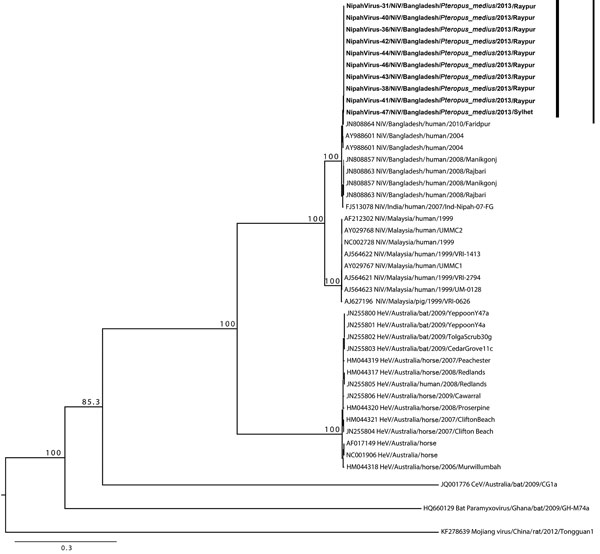
Figure 2. Phylogenetic tree of Nipah viruses from bats in Bangladesh (bold) compared with other henipaviruses, generated from full-genome sequences. Tree was constructed by using a maximum-likelihood approach, and robustness of nodes was tested with 1,000 bootstrap replicates. Sequences are labeled according to the following ordination: GenBank accession number or isolate identification number/virus type/country/host/year/strain. Numbers along branches are bootstrap values. Scale bar indicates nucleotide substitutions per site.
1These authors contributed equally to this article.
Page created: December 17, 2018
Page updated: December 17, 2018
Page reviewed: December 17, 2018
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.