Volume 25, Number 3—March 2019
Research
Mycobacterium avium in Community and Household Water, Suburban Philadelphia, Pennsylvania, USA, 2010–2012
Cite This Article
Citation for Media
Abstract
Attention to environmental sources of Mycobacterium avium complex (MAC) infection is a vital component of disease prevention and control. We investigated MAC colonization of household plumbing in suburban Philadelphia, Pennsylvania, USA. We used variable-number tandem-repeat genotyping and whole-genome sequencing with core genome single-nucleotide variant analysis to compare M. avium from household plumbing biofilms with M. avium isolates from patient respiratory specimens. M. avium was recovered from 30 (81.1%) of 37 households, including 19 (90.5%) of 21 M. avium patient households. For 11 (52.4%) of 21 patients with M. avium disease, isolates recovered from their respiratory and household samples were of the same genotype. Within the same community, 18 (85.7%) of 21 M. avium respiratory isolates genotypically matched household plumbing isolates. Six predominant genotypes were recovered across multiple households and respiratory specimens. M. avium colonizing municipal water and household plumbing may be a substantial source of MAC pulmonary infection.
Nontuberculous mycobacteria (NTM) are opportunistic human pathogens. Several species of NTM, including members of the Mycobacterium avium complex (MAC), can cause potentially life-threatening pulmonary infections that are difficult to treat (1,2). In 1989, Prince et al. described MAC pulmonary disease in persons without predisposing conditions (3). That study consisted of 21 patients from 2 hospitals in greater Philadelphia, Pennsylvania, USA, 1 of which was Lankenau Medical Center, located in Montgomery County. In 2012, Adjemian et al. identified Montgomery County as 1 of 7 US counties associated with a high risk for MAC lung disease (4). The reason for this risk was not apparent.
A recognized source of NTM infections is the environment (5–7). Many species of NTM are found in drinking water distribution systems (8), buildings (9), and household plumbing (7,10). Pulmonary NTM disease often recurs, even after completion of prolonged courses of therapy and periods of NTM-free sputum cultures (11). In a molecular epidemiologic study that confirmed transmission of M. avium from potable water to patients, the same strain of M. avium was found in 2 groups of patients with AIDS and in the recirculating hot water systems of the 2 hospitals at which they had been treated (12). To prevent NTM infection and reinfection of vulnerable populations, identification and elimination of environmental reservoirs is crucial. Remarkably, however, establishing epidemiologic links between clinical NTM isolates and specific environmental reservoirs has been difficult, partly because of the absence of robust tools for comparison of NTM isolates.
Newer molecular techniques, including variable-number tandem-repeat (VNTR) genotyping and whole-genome sequencing (WGS), provide greater discrimination of MAC isolates than previous methods (13–15). In this study, we used VNTR and WGS to test the hypothesis that household plumbing is a reservoir for NTM and is responsible for some cases of pulmonary MAC infection.
Study Setting and Population
Lankenau Medical Center is a community-based, academic medical center in Montgomery County, Pennsylvania, adjacent to Delaware and Philadelphia Counties. We prospectively identified and randomly chose study participants from patients at Lankenau Medical Center for whom MAC pulmonary infection was newly diagnosed during 2010–2012. Because the study was designed to investigate MAC pulmonary disease among women, we excluded male patients from the study. Chest computed tomography images were reviewed by the principal investigator (L.L.) and by a chest radiologist experienced in bronchiectasis (16). All patients had evidence of nodular disease and bronchiectasis. Potential participants for whom >2 sputum cultures or 1 bronchoscopic culture were positive for MAC were investigated further. Patients who met the microbiological, radiographic, and clinical criteria for MAC as outlined by the 2007 American Thoracic Society/Infectious Disease Society of America (ATS/IDSA) Statement on Nontuberculous Lung Disease (2) were offered study enrollment. Clinical and demographic information was obtained through chart review and from questionnaires that were filled out by patients with the aid of study personnel (Table 1).
The study protocol was approved by the Main Line Health Institutional Review Board, and informed consent was obtained from participants before study inclusion. Microbiology components of the study performed at partner institutions were deemed exempt from institutional review board review, and patient identifiers were removed from cultures or DNA samples before evaluation.
Control Population
Figure
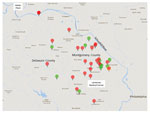
Figure. Area of study of Mycobacterium avium in community and household water, Philadelphia, Pennsylvania, USA, 2010–2012. The 26 patients (black tags) and 11 controls (gray tags) lived in suburban Philadelphia (Montgomery and...
For controls, we included 11 geographically matched persons from households serviced by the same municipal water system as the patients. Control participants were healthy neighbors of MAC patients and persons with bronchiectasis who had negative MAC culture results. All controls volunteered to have samples from their homes cultured (Figure).
Isolation and Identification of MAC
Isolation and identification of MAC from household biofilms were performed as described by Falkinham et al (6,7). We chose biofilm samples because the numbers of NTM are higher in biofilms than in water (7). Swab samples were vortexed in a tube for 60 s, then 0.1 mL of the suspended cells was spread in triplicate on Middlebrook 7H10 agar (Becton Dickinson and Company, https://www.bd.com) containing 0.5% glycerol and 10% oleic acid–albumin, sealed with Parafilm (Bemis NA, http://www.bemis.com), and incubated at 37°C. At weekly intervals, the plates were examined for evidence of mycobacterial colonies. Putative mycobacterial colonies were picked and streaked on M7H10 agar, and identical colonies on the original isolation M7H10 agar plates were counted to calculate CFUs per square centimeter of surface. After growth, colonies were acid-fast stained, DNA was isolated, and colonies were identified by PCR amplification and restriction digestion fragment pattern analysis of the hsp-65 gene. MAC were identified to species level by partial 16S rRNA gene sequencing, as previously described (13,17). M. avium isolates were subjected to PCR for IS901, and isolates negative for IS901 were classified as M. avium subsp. hominissuis (18,19).
Respiratory Samples
Respiratory samples were processed at the Lankenau Medical Center Microbiology Laboratory by use of standard methods, including a commercial hybridization assay (AccuProbe; Hologic, Inc., http://www.hologic.ca). For each patient for whom respiratory culture was positive, 1 MAC isolate was subcultured and sent for further analysis to the University of Texas Health Science Center (Tyler, TX, USA) and Virginia Polytechnic Institute and State University (Blacksburg, VA, USA) (Table 2). Species identification was performed by the same methods used for biofilm isolates.
Household Samples
Samples for mycobacterial culture were collected from all patient and control households (Table 2). Study personnel used sterile swabs to sample surfaces in contact with water, including kitchen plumbing (sink faucets, refrigerator ice and water dispensers), bathroom plumbing (shower pipes, showerheads, tub and sink faucets, toilet water tanks), and household humidifiers attached to central heating units. After sampling, each swab was placed in 3 mL of water from the source being tested, sealed in a sterile conical tube, and sent to the Falkinham Lab at Virginia Polytechnic Institute and State University. When cultures from a sample included colonies of diverse morphology, those colonies were counted, and a representative colony of each morphotype suspected of being an NTM was selected for analysis.
Genotyping of M. avium Isolates
VNTR genotyping of M. avium isolates was performed by using 6 previously characterized M. avium tandem-repeat sequences (MATR1, MATR2, MATR3, MATR7, MATR13, MATR14) (20) plus mycobacterial interspersed repetitive unit locus 3 (21). Internal transcribed spacer (ITS) sequencing (17,22) was also performed for all M. avium isolates. Results were compared with an in-house database that included genotyping results for 416 M. avium subsp. hominissuis isolates from 121 patients and 80 biofilm samples (18). Genotypes were assigned as previously published for M. avium subsp. hominissuis (13,18,23). New numbers were assigned to VNTR/ITS combinations not previously encountered (R.J. Wallace, Jr., unpub. data). VNTR 37a strains were further subtyped by using 3′ hsp65 sequencing (24) (Appendix Table 1).
Household Water Sources
We collected addresses for all participants and recorded them on a regional map (Figure). All patient and control households were located in southern Montgomery County or adjacent regions of Delaware County, Pennsylvania. According to the local water utility company, these areas of Montgomery and Delaware Counties are serviced by 1 water treatment plant, which processes surface water from the Schuylkill River and its tributaries (http://www.montcopa.org/DocumentCenter/View/4342). The age of the water pipes leading to the households ranged from 65 to 115 years. Most (65%) homes were >50 years old.
WGS
To assess genomic diversity within the most common VNTR types and the genetic similarity of respiratory and plumbing isolates from individual households, we performed WGS. We selected 40 M. avium isolates, representing genotypes 14a, 36, 22, and 37a (subtypes B1 and B2) (Appendix Table 2). Data associated with this study have been registered in the National Center for Biotechnology Information database as BioProject ID PRJNA339271 (https://www.ncbi.nlm.nih.gov/bioproject/339271). M. avium was isolated from the respiratory tract of 17 participants, from the respiratory tract and >1 household plumbing source for 12 of these 17, and from household plumbing only of the other 4 participants. A final participant was represented by 2 subcultures of a single respiratory isolate. These 2 biological replicates were processed separately and included to assess genomic variation that might be introduced during strain manipulation (e.g., subculture, DNA extraction, library preparation, and sequencing). Methods for WGS and bioinformatic analyses have been described (25). In brief, we generated paired-end libraries with the NEBNext Ultra DNA library prep kit and NEBNext Multiplex Oligos for Illumina (New England BioLabs, https://www.neb.ca). WGS was performed on an Illumina MiSeq platform by using the MiSeq reagent version 3 kit (600 cycle) according to the manufacturer’s guidelines (Illumina, Inc., https://www.illumina.com). High-quality core single-nucleotide polymorphism comparison was performed by using SNPhyl version 1.0.1 (26). Isolates from the same cluster were considered epidemiologically related if they differed by <15 single-nucleotide variants (SNVs) (27).
Patient Characteristics
All 26 patients were female, had nodular bronchiectasis confirmed by computed tomography, and had MAC lung disease as defined by the ATS/IDSA criteria (2). At enrollment, patient median age was 77 years (range 44–90 years), 97% (25/26) were white, 42% (11/26) were former smokers, and 88% (23/26) had no previous or co-occurring lung disease other than bronchiectasis (Table 1). All patients lived within 20 miles of each other (Figure): 20 in Montgomery County and 6 in Delaware County.
Control Characteristics
All 11 control participants were female; median age was 64 years (range 40–77 years). Eight lacked any history or signs of pulmonary NTM disease (i.e., no cough, dyspnea, fatigue, weight loss, or recurrent respiratory infections). Three participants had bronchiectasis and >2 sputum samples that did not grow NTM on culture. Controls lived within 20 miles of each other and the patients: 7 in Montgomery County and 4 in Delaware County (Figure).
Respiratory Samples
MAC respiratory isolates were recovered from 26 patients during the study period. The positive cultures were obtained from expectorated sputum and bronchoscopic sampling. We identified 3 species of MAC: M. avium subsp. hominissuis (21/26, 80.8%), M. intracellulare (3/26, 11.5%), and M. chimaera (2/26, 7.7%) (Table 2).
Household Samples
We collected 334 environmental biofilm samples, including 250 samples from 26 MAC patient households (range 4–19 samples from 3–13 sites in each household) and 84 samples from the 11 control households (range 3–14 samples from 3–11 sites in each household). A total of 95 M. avium isolates were recovered; 30/37 (81.1%) households were positive for M. avium, including 22/26 MAC patient households, 19/21 M. avium patient households, and 8/11 control households (Table 2).
M. avium was recovered from the kitchen sink faucet in 21/37 (56.8%) households and from 20/37 (54.1%) of the primary bathroom sites sampled, including 15/36 (41.7%) bathroom sink faucets, 13/35 (37.1%) showerheads, and 11/29 (37.9%) shower pipes (Appendix Table 3). Among households with humidifiers connected to the central heating units (with household plumbing as their water source), 7/17 (41.1%) were positive for M. avium. Among other sample sites, 2/14 (14.2%) refrigerator ice/water dispensers were positive, and 0 sampled toilet tanks were positive.
M. avium Genotypes
For 11/21 (52.4%) patients with M. avium disease, the genotype isolated from their respiratory sample was identical to the genotype recovered from their household plumbing sample (Table 2). For 18/21 (85.7%) M. avium patients, the respiratory isolate was the same genotype as that of plumbing biofilm isolates from >1 households within the same community. For 7/21 (33.3%) M. avium patients, the respiratory isolate genotype did not match that of a plumbing isolate from their own household but did match that of plumbing isolates from neighboring households. Overall, the 21 respiratory and 95 household isolates of M. avium encompassed 15 genotypes (Appendix Table 1). Six genotypes were broadly distributed across multiple households and respiratory specimens, and 2 genotypes were isolated from multiple household sources but not from respiratory specimens. In 11 households, the M. avium populations were heterogeneous, and 2–4 genotypes were recovered.
Whole-Genome Sequences and SNV–Based Phylogeny
SNVPhyl analysis of M. avium samples identified 26,871 variable sites across the core set of 4.3 million nt positions per isolate (Appendix Table 4). WGS was performed on 8 pairs of VNTR-matched respiratory and household isolates. For all 8 paired samples, the distance between respiratory and household isolates of M. avium was 4–51 SNVs; 5 pairs were separated by <15 SNVs (Table 3). The respiratory isolate from patient 13 differed from an isolate from her own kitchen sink by 37 SNVs and by <15 SNVs from plumbing biofilm isolates from households of patient 14 (9 SNVs) and patient 4 (11 SNVs). These patients did not know one another and had not visited each other’s homes.
M. avium patients 9, 10, and 11 resided in different apartments in the same high-rise building (Table 4). Across the 3 apartments, 32 sites were sampled and 17 M. avium cultures, representing 6 different genotypes, were recovered. The respiratory isolate from patient 9 was type 36, which was not found in her apartment but was the same as the respiratory isolate from patient 11 and was cultured from the apartment of patient 11. The respiratory isolate from patient 10 was type 22, which was the most common genotype recovered from her apartment (5/8 sites) and matched (i.e., 4 SNVs different) an isolate from her shower. Comparison of 5 type 14a strains isolated from sites across all 3 apartments revealed only minimal differences (25–63 SNVs). Detailed results of the WGS and SNVPhyl analyses are provided in the Appendix.
Determining the environmental source of infection for persons with pulmonary MAC disease has proven to be remarkably difficult. We used VNTR typing, targeted gene sequencing, and core-genome SNV analysis to compare M. avium isolates recovered from household plumbing biofilms with respiratory isolates from patients with MAC disease. We chose biofilm sampling of household plumbing, as opposed to direct water sampling, to enhance isolation rates of mycobacteria, which concentrate in biofilm. All households were serviced by the same water filtration plant and shared a common water source. MAC was widespread; 95 M. avium isolates were recovered from 22/26 (84.6%) MAC patient households (including 19/21 M. avium patient households) and 8/11 (72.7%) control households.
Respiratory and household isolates for 11/21 (52.4%) patients with M. avium nodular bronchiectatic pulmonary disease were genotypically matched. For 8 patients, the matched isolates were of the same genotype (14a). VNTR type 14a isolates were recovered from 10 (38.5%) respiratory and 16 (43.2%) household samples, and type 36 isolates were recovered from 4 (15.4%) respiratory and 10 (27%) household samples.
A 2016 report by Iakhaeva et al. (18) characterized 416 M. avium isolates from 120 patients and 80 environmental biofilms by using the methods that we used in this study and identified 49 VNTR types/subtypes. Since then, ≈100 additional isolates have undergone VNTR typing. Despite their predominance in our study, genotypes 14a and 36 were not identified in any previous studies (18; R.J. Wallace Jr., unpub. data). Our previous screening of additional patients and households in nearby areas that share the same primary water source (the Schuylkill River) but different water treatment plants found VNTR types 14a and 36 in those samples as well (R.J. Wallace Jr., unpub. data.)
VNTR genotyping is reproducible, portable, and discriminatory to the extent that isolates with different VNTR patterns are considered unrelated. However, as illustrated by the 37 and 37a subtypes found in this study, pseudoclustering can occur, such that isolates with identical VNTR patterns can have different ITS and hsp65 sequences (18). Previous work with M. tuberculosis also demonstrates that a VNTR match is not always proof of strain identity or an epidemiologic link (28,29). To better assess the relationship between VNTR-matched isolates, we turned to core-genome SNV analysis, which included ≈4.3 million nt positions; for phylogenetic comparisons, we used 26,871 variable sites. For the most common genotype identified in this study, type 14a, strains were split into 3 distinct subclusters (Appendix Figure). Isolates from the same subcluster were similar, differing by <100 of 26,871 SNVs, even though the sequenced strains were collected over 2 years and were recovered from diverse clinical and environmental sources. This widespread distribution and persistence is consistent with extensive colonization of the local water system with >1 M. avium strains endemic to Montgomery and Delaware Counties. For VNTR-matched respiratory and household isolates, the maximum distance was <60 SNVs. In 5 households, <15 SNVs separated the patient respiratory isolate from a plumbing biofilm isolate, suggesting an epidemiologic association between M. avium infection and household water. An additional patient respiratory isolate was <15 SNVs distant from biofilm strains recovered from neighboring households. This finding suggests that MAC exposure may occur outside the patient’s home but is more likely to represent incomplete recovery of respiratory strains, household strains, or both. Consistent with incomplete recovery, respiratory isolates from 7/21 (33.3%) M. avium patients had genotypes that were not present in their own households but did match isolates from neighboring households. Overall, 18/21 (85.7%) M. avium respiratory isolates matched >1 household biofilm isolates in the same community.
Our study had several limitations. The study population was relatively small and included only women from a specific region of Pennsylvania. To assess the universality of our findings, a larger, geographically diverse cohort is required. Because of the labor-intensive nature of the study, sampling was not comprehensive for all patients or households, and it is probable that because of undersampling some patient-household matches were missed. Longitudinal studies show that over time, patients with nodular bronchiectasis usually become infected with multiple genotypes of MAC (30), but we evaluated only 1 respiratory sample per patient.
The WGS component was conceived as a proof-of-principle experiment and limited to 40 isolates. The results of reference-based WGS comparisons are influenced by the analysis pipeline and choice of reference genome. Stringent pipeline filters intended to eliminate mapping errors can also suppress identification of true variants (31). Similarly, if a divergent genome is used as a reference, only a small subset of sequencing reads will be mapped and epidemiologically relevant SNVs may be missed (32). However, in our study, the core genome comprised >88% of the reference genome (≈4.3 million nt) despite the inclusion of genotypically diverse strains and the conservative filters used by the SNVPhyl pipeline.
Because an epidemiologic threshold for M. avium has not been definitively established, it remains unclear what SNV difference constitutes a definite genetic match. In a genomic analysis of M. abscessus, genetic relatedness was considered probable for isolates differing by <20 SNVs and possible for isolates differing by 20–38 SNVs (27). A cutoff of <12–25 nt for epidemiologically related isolates has been used in other studies of mycobacteria (33,34), but determining the most suitable threshold for M. avium will require additional isolates and analyses. In our WGS analysis of 8 pairs of VNTR-matched respiratory and household isolates, 5 pairs were separated by <15 SNVs, suggesting unequivocal genetic relatedness. In the other 3 pairs, distances were 24, 37, and 51 SNVs, which may also represent genetic relatedness but cannot be definitively established. In addition, the isolate from patient 13 was <15 SNVs distant from isolates from plumbing biofilm in the households of patients 14 (9 SNVs) and 4 (11 SNVs). These patients lived in separate towns that were 3–4 miles apart and serviced by the same water company, suggesting clonal infection from a common municipal water source. With MAC disease, there is often a delay between acquisition of infection and diagnosis, during which time mutations will accumulate in the environmental reservoir and the clinical isolates. For most cases in our study, infection probably preceded sample collection by several years. The estimated mutation rate for M. avium ssp. paratuberculosis is >0.5 substitutions/genome/year (35), but rates may be higher for M. avium subsp. hominissuis and have shorter doubling times. Rates may also be influenced by environmental and host factors. We recently sequenced serial isolates of M. intracellulare from patients receiving antimicrobial drug therapy and observed mutation rates that were 25-fold higher than for M. avium ssp. paratuberculosis (0.6–1 substitutions/mo) (25).
Our study identified household plumbing biofilms as a reservoir for M. avium and suggests that local rates of MAC lung disease may be influenced by mycobacterial colonization of municipal water. We cannot rule out other reservoirs, such as soil and dust, but thus far, recovery of M. avium from those sites and subsequent matches to patient isolates have been minimal (36). Moreover, because of innate resistance to chlorine and other disinfectants typically used in water treatment (37), M. avium has a survival advantage over most waterborne pathogens. In any geographic region, the prevalence of a particular Mycobacterium species probably affects disease rates. A recent study in Hawaii found that M. chimaera was the most common NTM recovered from household plumbing and the most common NTM recovered from patients in Hawaii (10). The value of understanding environmental sources of MAC infection have been highlighted by recent experiences with M. chimaera contamination of heater–cooler units (38).
Proof of an environmental source of M. avium has broad implications regarding prevention of recurrent infection in existing patients as well as prevention of new disease in susceptible persons. Additional studies are needed to establish effective methods for eliminating environmental reservoirs of M. avium and other problematic NTM. Tackling this challenging problem will require engagement of water utility workers, plumbers, environmental scientists, and engineers.
Dr. Lande is a clinical assistant professor at the Lankenau Institute for Medical Research, associate program director of the Pulmonary and Critical Care Fellowship Program at Lankenau Medical Center, and a clinical assistant professor of medicine at Thomas Jefferson University (Philadelphia, PA). Her main research interest is environmental sources of NTM.
Acknowledgment
Financial support for the Mycobacteria/Nocardia Research Laboratory was provided by in-house funding, patient donations, and the Amon G. Carter Foundation. Financial support for the household sampling was provided in part by the Lankenau Medical Center Women’s Board. Financial support for WGS was provided by the Saskatchewan Health Research Foundation, grant nos. 2867 and 3378.
References
- Marras TK, Daley CL. Epidemiology of human pulmonary infection with nontuberculous mycobacteria. Clin Chest Med. 2002;23:553–67. DOIPubMedGoogle Scholar
- Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, et al.; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367–416. DOIPubMedGoogle Scholar
- Prince DS, Peterson DD, Steiner RM, Gottlieb JE, Scott R, Israel HL, et al. Infection with Mycobacterium avium complex in patients without predisposing conditions. N Engl J Med. 1989;321:863–8. DOIPubMedGoogle Scholar
- Adjemian J, Olivier KN, Seitz AE, Falkinham JO III, Holland SM, Prevots DR. Spatial clusters of nontuberculous mycobacterial lung disease in the United States. Am J Respir Crit Care Med. 2012;186:553–8. DOIPubMedGoogle Scholar
- Falkinham JO III. Nontuberculous mycobacteria in the environment. Clin Chest Med. 2002;23:529–51. DOIPubMedGoogle Scholar
- Falkinham JO III, Iseman MD, de Haas P, van Soolingen D. Mycobacterium avium in a shower linked to pulmonary disease. J Water Health. 2008;6:209–13. DOIPubMedGoogle Scholar
- Falkinham JO III. Nontuberculous mycobacteria from household plumbing of patients with nontuberculous mycobacteria disease. Emerg Infect Dis. 2011;17:419–24. DOIPubMedGoogle Scholar
- Falkinham JO III, Norton CD, LeChevallier MW. Factors influencing numbers of Mycobacterium avium, Mycobacterium intracellulare, and other Mycobacteria in drinking water distribution systems. Appl Environ Microbiol. 2001;67:1225–31. DOIPubMedGoogle Scholar
- du Moulin GC, Stottmeier KD, Pelletier PA, Tsang AY, Hedley-Whyte J. Concentration of Mycobacterium avium by hospital hot water systems. JAMA. 1988;260:1599–601. DOIPubMedGoogle Scholar
- Honda JR, Hasan NA, Davidson RM, Williams MD, Epperson LE, Reynolds PR, et al. Environmental nontuberculous mycobacteria in the Hawaiian islands. PLoS Negl Trop Dis. 2016;10:e0005068. DOIPubMedGoogle Scholar
- Wallace RJ Jr, Zhang Y, Brown-Elliott BA, Yakrus MA, Wilson RW, Mann L, et al. Repeat positive cultures in Mycobacterium intracellulare lung disease after macrolide therapy represent new infections in patients with nodular bronchiectasis. J Infect Dis. 2002;186:266–73. DOIPubMedGoogle Scholar
- von Reyn CF, Maslow JN, Barber TW, Falkinham JO III, Arbeit RD. Persistent colonisation of potable water as a source of Mycobacterium avium infection in AIDS. Lancet. 1994;343:1137–41. DOIPubMedGoogle Scholar
- Iakhiaeva E, McNulty S, Brown Elliott BA, Falkinham JO III, Williams MD, Vasireddy R, et al. Mycobacterial interspersed repetitive-unit-variable-number tandem-repeat (MIRU-VNTR) genotyping of mycobacterium intracellulare for strain comparison with establishment of a PCR-based database. J Clin Microbiol. 2013;51:409–16. DOIPubMedGoogle Scholar
- Uchiya KI, Tomida S, Nakagawa T, Asahi S, Nikai T, Ogawa K. Comparative genome analyses of Mycobacterium avium reveal genomic features of its subspecies and strains that cause progression of pulmonary disease. Sci Rep. 2017;7:39750. DOIPubMedGoogle Scholar
- Ahlstrom C, Barkema HW, Stevenson K, Zadoks RN, Biek R, Kao R, et al. Genome-wide diversity and phylogeography of Mycobacterium avium subsp. paratuberculosis in Canadian dairy cattle. PLoS One. 2016;11:e0149017. DOIPubMedGoogle Scholar
- Wittram C, Weisbrod GL. Mycobacterium avium complex lung disease in immunocompetent patients: radiography-CT correlation. Br J Radiol. 2002;75:340–4. DOIPubMedGoogle Scholar
- Frothingham R, Wilson KH. Molecular phylogeny of the Mycobacterium avium complex demonstrates clinically meaningful divisions. J Infect Dis. 1994;169:305–12. DOIPubMedGoogle Scholar
- Iakhiaeva E, Howard ST, Brown Elliott BA, McNulty S, Newman KL, Falkinham JO III, et al. Variable-number tandem-repeat analysis of respiratory and household water biofilm isolates of “Mycobacterium avium subsp. hominissuis” with establishment of a PCR database. J Clin Microbiol. 2016;54:891–901. DOIPubMedGoogle Scholar
- Mijs W, de Haas P, Rossau R, Van der Laan T, Rigouts L, Portaels F, et al. Molecular evidence to support a proposal to reserve the designation Mycobacterium avium subsp. avium for bird-type isolates and ‘M. avium subsp. hominissuis’ for the human/porcine type of M. avium. Int J Syst Evol Microbiol. 2002;52:1505–18.PubMedGoogle Scholar
- Inagaki T, Nishimori K, Yagi T, Ichikawa K, Moriyama M, Nakagawa T, et al. Comparison of a variable-number tandem-repeat (VNTR) method for typing Mycobacterium avium with mycobacterial interspersed repetitive-unit-VNTR and IS1245 restriction fragment length polymorphism typing. J Clin Microbiol. 2009;47:2156–64. DOIPubMedGoogle Scholar
- Bull TJ, Sidi-Boumedine K, McMinn EJ, Stevenson K, Pickup R, Hermon-Taylor J. Mycobacterial interspersed repetitive units (MIRU) differentiate Mycobacterium avium subspecies paratuberculosis from other species of the Mycobacterium avium complex. Mol Cell Probes. 2003;17:157–64. DOIPubMedGoogle Scholar
- Dauchy FA, Dégrange S, Charron A, Dupon M, Xin Y, Bébéar C, et al. Variable-number tandem-repeat markers for typing Mycobacterium intracellulare strains isolated in humans. BMC Microbiol. 2010;10:93. DOIPubMedGoogle Scholar
- Stout JE, Hopkins GW, McDonald JR, Quinn A, Hamilton CD, Reller LB, et al. Association between 16S-23S internal transcribed spacer sequence groups of Mycobacterium avium complex and pulmonary disease. J Clin Microbiol. 2008;46:2790–3. DOIPubMedGoogle Scholar
- Turenne CY, Semret M, Cousins DV, Collins DM, Behr MA. Sequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complex. J Clin Microbiol. 2006;44:433–40. DOIPubMedGoogle Scholar
- Alexander DC, Vasireddy R, Vasireddy S, Philley JV, Brown-Elliott BA, Perry BJ, et al. Emergence of mmpT5 variants during bedaquiline treatment of Mycobacterium intracellulare lung disease. J Clin Microbiol. 2017;55:574–84. DOIPubMedGoogle Scholar
- Petkau A, Mabon P, Sieffert C, Knox NC, Cabral J, Iskander M, et al. SNVPhyl: a single nucleotide variant phylogenomics pipeline for microbial genomic epidemiology. Microb Genom. 2017;3:e000116. DOIGoogle Scholar
- Bryant J, Grogono D, Rodriguez-Rinco D, Everall I, Brown KP, Moreno P, et al. Population-level genomics identifies the emergence and global spread of a human transmissible multidrug resistant nontuberculous mycobacterium. Science. 2016;354:751–7. DOIPubMedGoogle Scholar
- Jamieson FB, Teatero S, Guthrie JL, Neemuchwala A, Fittipaldi N, Mehaffy C. Whole-genome sequencing of the Mycobacterium tuberculosis Manila sublineage results in less clustering and better resolution than mycobacterial interspersed repetitive-unit-variable-number tandem-repeat (MIRU-VNTR) typing and spoligotyping. J Clin Microbiol. 2014;52:3795–8. DOIPubMedGoogle Scholar
- Mehaffy C, Guthrie JL, Alexander DC, Stuart R, Rea E, Jamieson FB. Marked microevolution of a unique Mycobacterium tuberculosis strain in 17 years of ongoing transmission in a high risk population. PLoS One. 2014;9:e112928. DOIPubMedGoogle Scholar
- Wallace RJ Jr, Zhang Y, Brown BA, Dawson D, Murphy DT, Wilson R, et al. Polyclonal Mycobacterium avium complex infections in patients with nodular bronchiectasis. Am J Respir Crit Care Med. 1998;158:1235–44. DOIPubMedGoogle Scholar
- Guthrie JL, Gardy JL. A brief primer on genomic epidemiology: lessons learned from Mycobacterium tuberculosis. Ann N Y Acad Sci. 2017;1388:59–77. DOIPubMedGoogle Scholar
- Lee RS, Behr MA. Does choice matter? Reference-based alignment for molecular epidemiology of tuberculosis. J Clin Microbiol. 2016;54:1891–5. DOIPubMedGoogle Scholar
- Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet. 2013;381:1551–60. DOIPubMedGoogle Scholar
- Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–46. DOIPubMedGoogle Scholar
- Bryant JM, Thibault VC, Smith DG, McLuckie J, Heron I, Sevilla IA, et al. Phylogenomic exploration of the relationships between strains of Mycobacterium avium subspecies paratuberculosis. BMC Genomics. 2016;17:79. DOIPubMedGoogle Scholar
- Fujita K, Ito Y, Hirai T, Maekawa K, Imai S, Tatsumi S, et al. Genetic relatedness of Mycobacterium avium-intracellulare complex isolates from patients with pulmonary MAC disease and their residential soils. Clin Microbiol Infect. 2013;19:537–41. DOIPubMedGoogle Scholar
- Taylor RH, Falkinham JO III, Norton CD, LeChevallier MW. Chlorine, chloramine, chlorine dioxide, and ozone susceptibility of Mycobacterium avium. Appl Environ Microbiol. 2000;66:1702–5. DOIPubMedGoogle Scholar
- Schreiber PW, Kuster SP, Hasse B, Bayard C, Rüegg C, Kohler P, et al. Reemergence of Mycobacterium chimaera in heater-cooler units despite intensified cleaning and disinfection protocol. Emerg Infect Dis. 2016;22:1830–3. DOIPubMedGoogle Scholar
Figure
Tables
Cite This ArticleOriginal Publication Date: February 19, 2019
Table of Contents – Volume 25, Number 3—March 2019
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Leah Lande, Lankenau Medical Center and Lankenau Institute for Medical Research, Division of Pulmonary and Critical Care Medicine, 100 Lancaster Ave, MOB West, Ste 230, Wynnewood, PA 19096, USA
Top