Volume 25, Number 3—March 2019
Dispatch
Prospective Whole-Genome Sequencing in Tuberculosis Outbreak Investigation, France, 2017–2018
Cite This Article
Citation for Media
Abstract
During June 2017–April 2018, active tuberculosis with Beijing SIT1 isolates was diagnosed in 14 persons living in 4 distant cities in France. Whole-genome sequencing indicated that these patients belonged to a single transmission chain. Whole-genome sequencing–based laboratory investigations enabled prompt tracing of linked cases to improve tuberculosis control.
France is a low-prevalence country for tuberculosis (TB); mean incidence was 7.1 cases/100,000 inhabitants in 2015 (1). Auvergne-Rhône-Alpes is the region of France with the second highest prevalence of TB (428 cases notified in 2015). Genotyping is not yet routinely performed for all Mycobacterium tuberculosis isolates in France. However, prospective surveys of M. tuberculosis strains isolated in the Auvergne-Rhône-Alpes region have been conducted since 2008 by using systematic genotyping to detect TB transmission events and to comprehend complex relationships between certain lineages and the geographic origin of clinical cases (approved by the Comité de Protection des Personnes Sud-Est IV under no. DC-2011–1306) (2,3). Since November 2016, whole-genome sequencing (WGS) of M. tuberculosis isolates was implemented in the Lyon University Hospital laboratory (Lyon, France) on a routine basis to discriminate transmission clusters otherwise undetected by the spoligotyping method. We previously reported on the effect of immigration on imported M. tuberculosis cases in the Auvergne-Rhône-Alpes region (3); here, we report the rapid spread of a specific M. tuberculosis strain and the need for continued vigilant surveillance using advanced molecular techniques.
During June 2017–April 2018, active TB was diagnosed in 11 persons living in the same city of Auvergne-Rhône-Alpes; all isolates belonged to Beijing SIT1 spoligotype. WGS analysis confirmed that these 11 patients were infected with the same strain, along with 3 other isolates collected in 3 different and distant cities in France, indicating a clonal outbreak.
Figure 1
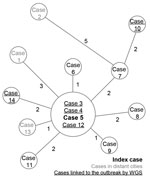
Figure 1. Minimum-spanning tree of single-nucleotide polymorphism differences from 14 Mycobacterium tuberculosis outbreak isolates, France, 2017–2018. Cases were numbered according to sampling chronology. Sizes of circles are proportional to the number of isolates...
During June–September 2017, three cases of active TB were diagnosed from the same city in the Auvergne-Rhône-Alpes region. Patients belonged to 2 families without previously known relationships. One of the 3 patients (index case-patient [chronological case-patient 5]) had a 2-year history of confirmed TB; this patient originated from Cape Verde and reported frequent recent travel there. Direct microscopic examination of sputum showed very high titers of acid-fast bacilli, implying a high risk for onward M. tuberculosis transmission. In the other 2 case-patients (chronological case-patients 3 and 4), TB was newly diagnosed; they had no history of or suspicion for infection, but both resided in the same household. Isolates from all 3 cases were susceptible to standard first-line anti-TB drugs, and spoligotyping revealed that all belonged to Beijing SIT1 lineage. In parallel with conventional membrane-based spoligotyping, WGS was performed to ascertain whether it was the same strain (Appendix). WGS analysis indicated that the strains were identical, meaning 0 single-nucleotide polymorphism (SNP) distance (Figure 1).
Investigations and interviews conducted by the TB control center later revealed that the 2 families did have a connection, through frequent contacts between case-patient 4 and the index case-patient (case-patient 5). These results led the TB control center to widen the circle of investigations beyond household and frequent contacts and to expand the investigation to occasional contacts (including persons from other geographic regions) and persons attending the public places frequented by the index case-patient.
During September 2017–April 2018, extended contact tracing identified 5 additional cases of active TB within the index city and 3 epidemiologically linked cases in distant areas (Île-de-France, Hauts-de-France, and Provence-Alpes-Côte d’Azur). WGS analysis confirmed that all patients belonged to a single transmission chain, supporting a rapid dissemination of this epidemic strain in France. TB had been diagnosed in 1 of the in-contact case-patients as early as 2015; case-patient 1 and the index case-patient lived together in 2015 in Île-de-France, during which time the transmission event was hypothesized to have occurred. These findings led to an extended investigation and communication outreach to general practitioners in the index city. General practitioners were informed of the outbreak and prompted to prioritize performing TB detection in patients with respiratory symptoms suspected to be TB. Consequently, during early 2018, active TB caused by the Beijing SIT1 epidemic strain was diagnosed in 3 previously non–epidemiologically linked patients. Subsequent investigations showed previously missed occasional contacts between the index case-patient and these 3 TB case-patients.
Out of the 14 case-patients identified, 9 (64%) infected by the Beijing strain were household or frequent contacts, and 5 (36%) were occasional contacts (Table). Five case-patients were children. Pulmonary and extrapulmonary disease developed in 4 case-patients; in 2 of those, meningitis, a severe form of TB associated with high rates of death and disability, developed.
Among the 14 cases identified, 5 (36%) were linked to this outbreak through laboratory investigations. WGS analysis enabled us to link strains from 2 initially independent contact tracing investigations and relate 5 cases with no previously identified epidemiologic link. Pairwise SNP distance comparison of the isolates revealed a maximum of 7 SNPs separating the index case-patient from the other 13 case-patients and a maximum of 10 SNPs separating other cases (Figure 1). The median SNP difference between cases with a verified epidemiologic link was 2 (range 0–5). This finding is consistent with previous studies in which a 12-SNP threshold excluded transmission (4). We compared the cluster we report with other Beijing SIT1 strains isolated in 2017 in our region: pairwise SNP comparison ranged from 140- to 330-SNP distance. This cluster is therefore unrelated to other Beijing SIT1 contemporarily and regionally circulating strains. According to the Shitikov scheme (5), those strains belong to the Central Asia group.
Figure 2

Figure 2. Prevalence of tuberculosis cases and Mycobacterium tuberculosis Beijing SIT1 genotype, Auvergne-Rhône-Alpes, France, 2008–2018. DS, drug-sensitive; MDR, multidrug-resistant.
The Beijing strain family, regardless of the antimicrobial susceptibility profile, has been involved in many TB outbreaks, including in Europe (6–10). Some studies suggest that the epidemic success of the modern Beijing strains might be linked to increased virulence, higher transmission rates, or both, but findings have not been consistent (11–13). In the Auvergne-Rhône-Alpes region, Beijing strain accounts for 2.5%–8.1% of all M. tuberculosis isolates (average 4.9%) (Figure 2), depending on the year. The increased incidence of Beijing genotype during 2011–2013 (i.e., up to 7.3% in 2012) was related to the migratory influx of patients with multidrug-resistant TB from Eastern Europe and Asia (14). The 8.1% increased incidence of Beijing genotype from the second half of 2017 and the first 4 months of 2018 was due to the outbreak we describe.
WGS led to the rapid identification of this transmission event. Real-time feedback between the microbiology laboratory and the TB control center prompted further contact investigations and identification of additional epidemiologically linked cases.
This report highlights how, beyond the traditional contact tracing investigations, molecular M. tuberculosis typing enabled detection of otherwise unsuspected transmission and therefore identification of extended clusters. This finding is important because it is not uncommon that TB patients do not cooperate well with interviews (15); even in low-prevalence countries, stigma associated with TB diagnosis often hampers adherence of patients to screening. Noncomplying patients do not disclose exhaustive lists of contacts; as shown in this study, case-patients 4 and 5 were found to have frequent contact only through the second contact-tracing interview. In this respect, robust microbiological evidence of transmission may encourage TB control units to perform second interviews and thus increase the chances to document more transmission events. Our observations underscore the added value of molecular epidemiologic tools to prompt TB contact investigations for better detection and disease control.
Dr. Genestet is a postdoctoral fellow at Centre International de Recherche en Infectiologie in Lyon, France. Her research interests include M. tuberculosis epidemiology and discovery of genetic markers associated with physiopathology of tuberculosis.
Acknowledgment
We thank the French National Reference Center for Mycobacteria and Antimycobacterial Resistance for fruitful discussions. We also thank Krystian Valenducq for contributing bioinformatics expertise.
References
- Guthmann J-P, Aït Belghiti F, Lévy-Bruhl D. Tuberculosis epidemiology in France in 2015. Impact of the suspension of BCG mandatory vaccination on child tuberculosis, 2007–2015. Bull Epidémiol Hebd. 2017 [cited 2018 Dec 17]. http://invs.santepubliquefrance.fr/beh/2017/7/2017_7_1.html
- Barbier M, Dumitrescu O, Pichat C, Carret G, Ronnaux-Baron A-S, Blasquez G, et al. Changing patterns of human migrations shaped the global population structure of Mycobacterium tuberculosis in France. Sci Rep. 2018;8:5855. DOIPubMedGoogle Scholar
- Pichat C, Couvin D, Carret G, Frédénucci I, Jacomo V, Carricajo A, et al. Combined genotypic, phylogenetic, and epidemiologic analyses of Mycobacterium tuberculosis genetic diversity in the Rhône Alpes region, France. PLoS One. 2016;11:e0153580. DOIPubMedGoogle Scholar
- Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–46. DOIPubMedGoogle Scholar
- Shitikov E, Kolchenko S, Mokrousov I, Bespyatykh J, Ischenko D, Ilina E, et al. Evolutionary pathway analysis and unified classification of East Asian lineage of Mycobacterium tuberculosis. Sci Rep. 2017;7:9227. DOIPubMedGoogle Scholar
- Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. 2015;47:242–9. DOIPubMedGoogle Scholar
- Caminero JA, Pena MJ, Campos-Herrero MI, Rodríguez JC, García I, Cabrera P, et al. Epidemiological evidence of the spread of a Mycobacterium tuberculosis strain of the Beijing genotype on Gran Canaria Island. Am J Respir Crit Care Med. 2001;164:1165–70. DOIPubMedGoogle Scholar
- Faccini M, Codecasa LR, Ciconali G, Cammarata S, Borriello CR, De Gioia C, et al. Tuberculosis outbreak in a primary school, Milan, Italy. Emerg Infect Dis. 2013;19:485–7. DOIPubMedGoogle Scholar
- Golesi F, Brignatz J, Bellenfant M, Raoult D, Drancourt M. Mycobacterium tuberculosis Beijing outbreak in a school in Marseille, France, 2012. Euro Surveill. 2013;18:20354.PubMedGoogle Scholar
- Norheim G, Seterelv S, Arnesen TM, Mengshoel AT, Tønjum T, Rønning JO, et al. Tuberculosis outbreak in an educational institution in Norway. J Clin Microbiol. 2017;55:1327–33. DOIPubMedGoogle Scholar
- Hanekom M, Gey van Pittius NC, McEvoy C, Victor TC, Van Helden PD, Warren RM. Mycobacterium tuberculosis Beijing genotype: a template for success. Tuberculosis (Edinb). 2011;91:510–23. DOIPubMedGoogle Scholar
- Parwati I, van Crevel R, van Soolingen D. Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. Lancet Infect Dis. 2010;10:103–11. DOIPubMedGoogle Scholar
- Ribeiro SCM, Gomes LL, Amaral EP, Andrade MRM, Almeida FM, Rezende AL, et al. Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J Clin Microbiol. 2014;52:2615–24. DOIPubMedGoogle Scholar
- Berland J-L, Genestet C, Ginevra C, Claude J-B, Pichat C, Hodille E, et al. Cross-transmission of multi-drug resistant tuberculosis (MDR-TB) in a low-prevalence setting explored by whole genome sequencing (WGS). Presented at: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017 Apr 22–25; Vienna, Austria.
- Craig GM, Daftary A, Engel N, O’Driscoll S, Ioannaki A. Tuberculosis stigma as a social determinant of health: a systematic mapping review of research in low incidence countries. Int J Infect Dis. 2017;56:90–100. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 25, Number 3—March 2019
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Charlotte Genestet, Centre International de Recherche en Infectiologie, Fondation Mérieux, 21 avenue Tony Garnier, 69007 Lyon, France
Top