Volume 25, Number 4—April 2019
Dispatch
Genomic Survey of Bordetella pertussis Diversity, United States, 2000–2013
Cite This Article
Citation for Media
Abstract
We characterized 170 complete genome assemblies from clinical Bordetella pertussis isolates representing geographic and temporal diversity in the United States. These data capture genotypic shifts, including increased pertactin deficiency, occurring amid the current pertussis disease resurgence and provide a foundation for needed research to direct future public health control strategies.
Whooping cough (pertussis) remains a public health challenge in the United States where, despite high vaccine coverage, an increased number of cases have been reported since the late 1980s. This resurgence has included >48,000 cases reported in 2012 and notable recent statewide epidemics (1). Likely causes of the increase in reporting include heightened awareness, expanded surveillance, improved laboratory diagnostics, and waning protection conferred by acellular pertussis (aP) vaccine formulations (1,2).
The United States exclusively uses aP vaccines composed of inactivated Bordetella pertussis immunogenic proteins pertussis toxin (Pt), pertactin (Prn), and filamentous hemagglutinin (Fha), either with or without fimbria (Fim) types 2 and 3. Genetic divergence of circulating B. pertussis away from vaccine reference strains has led to allelic mismatch and the rapid emergence of Prn deficiency (3). Although such recent genetic changes may be ascribed to vaccine-driven immune selection (4), aP vaccines remain effective (5).
The chromosome of B. pertussis also undergoes frequent structural rearrangement (6) that presents unique challenges to thorough investigation of genetic contributions to disease resurgence, limiting assessment of public health strategies. Until recently, genomic data with sufficient resolution to study sequence and structural variation were available only for vaccine and laboratory reference strains. However, pathogen evolution must be explored through multinomic characterization of circulating genotypes. To address this gap, we developed a dataset of complete, reference-quality genome sequence assemblies from isolates representing the geographic and temporal diversity of B. pertussis circulating in the United States during 2000–2013.
Figure 1
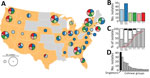
Figure 1. Bordetella pertussis diversity, United States, 2000–2013. A) Geographic origin of B. pertussis isolates selected to maximize the number of source states from each of 6 time periods. Pie chart diameter represents...
The Centers for Disease Control and Prevention (CDC) maintains a collection of B. pertussis isolates recovered by state public health laboratories through routine surveillance and outbreaks or the Enhanced Pertussis Surveillance/Emerging Infections Program Network (7). We selected a subset of isolates (n = 170) to account for potential geographic diversity. We stratified all isolates in the collection by state and time period (2000–2002, 2003–2009, 2010, 2011, 2012, and 2013) chosen according to diversity indices reported previously (8), with additional emphasis on more recent sampling. We then randomly sampled the stratified collection to maximize the number of source states (n = 34) during each period with equal weighting (Figure 1, panel A, B). Most isolates were characterized by existing molecular approaches, multilocus variable-number tandem-repeat analysis (MLVA), and pulsed-field gel electrophoresis (PFGE), as described previously (9). The selected isolates included 17 MLVA types, with type 27 the most prevalent, and 33 PFGE profiles, with profile CDC013 the most prevalent (Appendix Table 1).
We performed whole-genome shotgun sequencing and assembly as described previously (10) (Appendix). Genome assembly yielded a single circular contig for all isolates, and we performed sequence-based molecular typing (Appendix). Nearly all isolates (96%) were of the predominant type prn2-ptxP3-ptxA1 with either fimH1 or fimH2, and few harbored alternate types such as prn1-ptxP1-ptxA2-fimH1 (Figure 1, panel C). Prn deficiency has been observed in >16 independent mutations to prn (6); we observed 10 deficient alleles among 57/170 isolates in our study, including missense substitutions, deletions, promoter disruption, and various IS481 insertions. The proportion of isolates with Prn-deficient alleles increased rapidly beginning in 2010 (Figure 1, panel C), consistent with a larger molecular survey of US isolates conducted previously that included some used in this study (3). We also determined MLVA type from genome assemblies using a custom bioinformatics pipeline (wgsMLVA) based on traditional PCR primer sequences (11) (Appendix). None of the genomes encoded known 23S ribosomal RNA mutation associated with erythromycin resistance (12).
To determine variation in chromosome structure, we performed exhaustive pairwise alignment of assembled genomes as previously described (6). Of the 170 assemblies, 129 clustered into 16 groups of >2 colinear genomes (lacking observable rearrangement or deletion >1,500 bp), whereas 41 assemblies (singletons) exhibited unique structures not shared with any others in the dataset. Observed structures largely correlated with PFGE, a proxy for chromosome structure, clustering isolates with shared PFGE profiles. The abundance of common structures reflected predominant PFGE profiles, and the largest cluster corresponded to profile CDC013 (Figure 1, panel D). Differences between many common structures could be attributed to large inversions flanked by insertions of the multicopy IS481. Select singleton structures resulted from tandem duplication of large regions (15.5–190 kbp) in the genomes of 5 isolates (D236, D665, H624, J085, and J139) that were also flanked by copies of IS481.
Figure 2

Figure 2. Phylogenetic reconstruction of all 170 isolates and the reference Tohama I (GenBank accession no. CP010964). Isolate metadata and molecular characteristics are color coded, as detailed in the key. Scale bar indicates...
We reconstructed a maximum-likelihood phylogeny of the isolate genomes from 840 core variable single-nucleotide polymorphisms (SNPs) determined from the reference Tohama I (GenBank accession no. CP010964) (Appendix). The resulting tree topology revealed deep divergence of lineages bearing alleles ptxP1 and ptxP3, as well as clear distinctions between clades of prn2-ptxP3-ptxA1-fimH1 and prn2-ptxP3-ptxA1-fimH2 (Figure 2). Only certain prn-disrupting mutations (e.g., nonsense C1273T, promoter disruption) and chromosome structures (e.g., cluster-4, cluster-6, cluster-7, cluster-9) appeared phylogenetically linked, meaning isolates sharing them were also related according to their SNP patterns. However, each group of related isolates was recovered across multiple states and time periods, suggesting that genotypes, whether defined by gene sequence or chromosome structure, were stable enough to be widely circulated. Prn deficiency due to IS481 disruption has resulted from >7 independent events among the isolates in this dataset, but related isolates with these mutations were likewise geographically and temporally distributed. These results are consistent with phylogenies of circulating B. pertussis reported elsewhere (6,13).
We have developed a representative dataset of complete genome sequence assemblies derived from B. pertussis clinical isolates recovered in the United States that captures shifting population genetics concurrent with disease resurgence. We selected isolates to maximize the geographic diversity of circulating B. pertussis across 6 time periods during 2000–2013 and to span the time period in which Prn deficiency emerged as the predominant molecular type. Although the sparse sampling of individual states and regions prohibited detailed analyses of geographic distribution, we did recover isolates with shared SNP patterns and chromosome structures from disparate states. Our results illustrate underlying challenges to the molecular study of pertussis resurgence, including a circulating mixture of gene sequence (SNP) and chromosome structure variants.
The genomic data we provide will aid open research toward improved vaccine development and disease control strategies. Because little to no such high-quality data existed previously, the contribution of genome evolution to pertussis resurgence has not been fully appreciated. A subset of these data has already helped elucidate historical patterns of chromosome rearrangement (6). However, comparative genomics alone is not sufficient to understand the resurgence in pertussis. Further laboratory experimentation using in vitro and in vivo infection models is needed to link outcomes with novel, bioinformatically determined genetic variation, such as discrete rearrangements and tandem duplications. Potential differences in antigen expression resulting from these changes in gene organization, which may influence the burden of disease, remain untested. Our results provide needed context to guide such investigations by highlighting representative, circulating genotypes as they continue their divergence from existing laboratory and vaccine reference strains. Data such as those presented here critically establish the necessary foundation for collaborative development of advanced diagnostics, novel molecular typing methods, and improved vaccine formulations.
Dr. Weigand is a bioinformatics research scientist in the Pertussis and Diphtheria Laboratory, Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases, CDC, Atlanta. His primary research interest is comparative genomics of bacterial pathogens, with a current focus on Bordetella pertussis.
Acknowledgments
Additional members of the CDC Pertussis Working Group: Dhwani Batra, Katherine E. Bowden, Mark Burroughs, Pamela K. Cassiday, Jamie K. Davis, Taccara Johnson, Hong Ju, Phalasy Juieng, Kristen Knipe, Vladimir N. Loparev, Stacey W. Martin, Christine Miner, Lori A. Rowe, Tami H. Skoff, Mili Sheth, Kevin Tang.
This work was made possible through support from the Advanced Molecular Detection program at the US Centers for Disease Control and Prevention.
References
- Clark TA. Changing pertussis epidemiology: everything old is new again. J Infect Dis. 2014;209:978–81. DOIPubMedGoogle Scholar
- Ausiello CM, Cassone A. Acellular pertussis vaccines and pertussis resurgence: revise or replace? MBio. 2014;5:e01339–14. DOIPubMedGoogle Scholar
- Pawloski LC, Queenan AM, Cassiday PK, Lynch AS, Harrison MJ, Shang W, et al. Prevalence and molecular characterization of pertactin-deficient Bordetella pertussis in the United States. Clin Vaccine Immunol. 2014;21:119–25. DOIPubMedGoogle Scholar
- Martin SW, Pawloski L, Williams M, Weening K, DeBolt C, Qin X, et al. Pertactin-negative Bordetella pertussis strains: evidence for a possible selective advantage. Clin Infect Dis. 2015;60:223–7. DOIPubMedGoogle Scholar
- Breakwell L, Kelso P, Finley C, Schoenfeld S, Goode B, Misegades LK, et al. Pertussis vaccine effectiveness in the setting of pertactin-deficient pertussis. Pediatrics. 2016;137:e20153973. DOIPubMedGoogle Scholar
- Weigand MR, Peng Y, Loparev V, Batra D, Bowden KE, Burroughs M, et al. The history of Bordetella pertussis genome evolution includes structural rearrangement. J Bacteriol. 2017;199:e00806–16. DOIPubMedGoogle Scholar
- Skoff TH, Baumbach J, Cieslak PR. Tracking pertussis and evaluating control measures through enhanced pertussis surveillance, Emerging Infections Program, United States. Emerg Infect Dis. 2015;21:1568–73. DOIPubMedGoogle Scholar
- Schmidtke AJ, Boney KO, Martin SW, Skoff TH, Tondella ML, Tatti KM. Population diversity among Bordetella pertussis isolates, United States, 1935-2009. Emerg Infect Dis. 2012;18:1248–55. DOIPubMedGoogle Scholar
- Hardwick TH, Cassiday P, Weyant RS, Bisgard KM, Sanden GN. Changes in predominance and diversity of genomic subtypes of Bordetella pertussis isolated in the United States, 1935 to 1999. [_article]. Emerg Infect Dis. 2002;8:44–9. DOIPubMedGoogle Scholar
- Bowden KE, Weigand MR, Peng Y, Cassiday PK, Sammons S, Knipe K, et al. Genome structural diversity among 31 Bordetella pertussis isolates from two recent U.S. whooping cough statewide epidemics. mSphere. 2016;May–Jun:e00036–16. DOIGoogle Scholar
- Schouls LM, van der Heide HG, Vauterin L, Vauterin P, Mooi FR. Multiple-locus variable-number tandem repeat analysis of Dutch Bordetella pertussis strains reveals rapid genetic changes with clonal expansion during the late 1990s. J Bacteriol. 2004;186:5496–505. DOIPubMedGoogle Scholar
- Bartkus JM, Juni BA, Ehresmann K, Miller CA, Sanden GN, Cassiday PK, et al. Identification of a mutation associated with erythromycin resistance in Bordetella pertussis: implications for surveillance of antimicrobial resistance. J Clin Microbiol. 2003;41:1167–72. DOIPubMedGoogle Scholar
- Bart MJ, Harris SR, Advani A, Arakawa Y, Bottero D, Bouchez V, et al. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. MBio. 2014;5:e01074. DOIPubMedGoogle Scholar
Figures
Cite This ArticleOriginal Publication Date: March 13, 2019
1Additional members of the CDC Pertussis Working Group are listed at the end of this article.
Table of Contents – Volume 25, Number 4—April 2019
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Michael R. Weigand, Centers for Disease Control and Prevention, 1600 Clifton Rd NE, Mailstop H18-B, Atlanta, GA 30329-4027, USA
Top