Volume 25, Number 7—July 2019
Research Letter
Crimean-Congo Hemorrhagic Fever Virus Genome in Tick from Migratory Bird, Italy
Figure
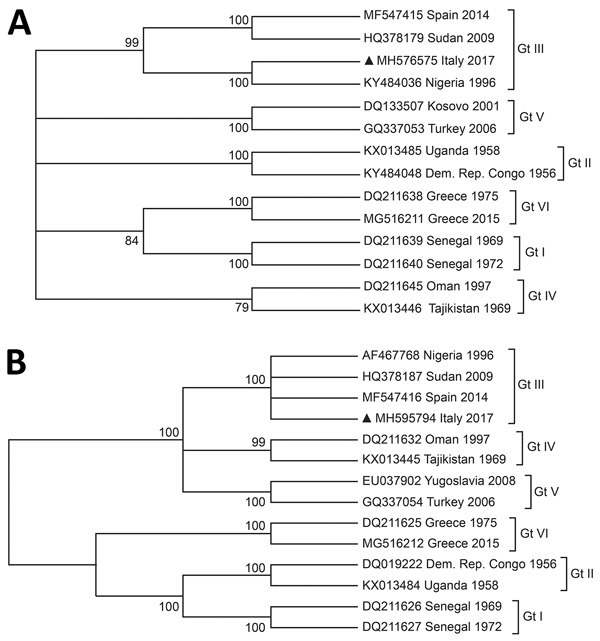
Figure. Cladistic relationship of Crimean-Congo hemorrhagic fever virus gene found in a Hyalomma rufipes nymph collected from a whinchat (Saxicola rubetra) in Italy (black triangles) with strains representative of the circulating genotypes. The analysis is based on partial sequences of the small segment (A) and medium segment (B) of the virus genomes. We used the neighbor-joining method based on the Tamura-Nei model. Percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The analysis involved 14 nucleotide sequences. Codon positions included were first, second, third, and noncoding. All positions containing gaps and missing data have been eliminated. A total of 485 positions (small segment) and 1,374 positions (medium segment) were included in the final dataset. GenBank accession numbers are provided. Gt, genotype.