Volume 26, Number 12—December 2020
Research
Human-Pathogenic Kasokero Virus in Field-Collected Ticks
Figure 2
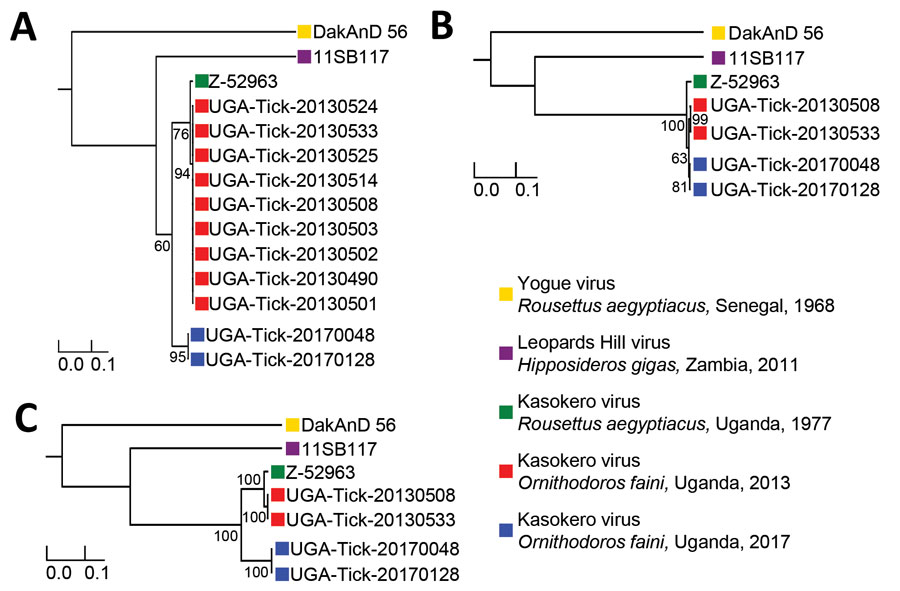
Figure 2. Maximum-likelihood phylogenies constructed from virus sequences belonging to the species Kasokero orthonairovirus, including viruses collected from Ornithodoros (Reticulinasus) faini tick pools from Rousettus aegyptiacus bats, western Uganda, 2013 and 2017. The midpoint rooted phylogenies were generated from complete nucleoprotein (N) (A), glycoprotein precursor (GP) (B), and RNA-dependent RNA-polymerase (RdRp) (C) gene sequences. The N and RdRp gene phylogenies were generated using the general time-reversible nucleotide substitution model with the addition of invariant sites, and the GP gene phylogeny was generated using the general time-reversible nucleotide substitution model with a gamma distribution of rates across sites. Horizontal branch lengths are proportional to the genetic distance between the sequences. Numbers at the end of the branches represent percent bootstrap values based on 1,000 replicates. Only percent bootstrap values >50% are shown. GenBank accession numbers for the new Kasokero virus tick sequences from this study are MT309080–98. Scale bars indicate nucleotide substitutions per site.