Volume 26, Number 5—May 2020
Research
Rhizopus microsporus Infections Associated with Surgical Procedures, Argentina, 2006–2014
Cite This Article
Citation for Media
Abstract
Rhizopus spp. fungi are ubiquitous in the environment and a rare but substantial cause of infection in immunosuppressed persons and surgery patients. During 2005–2017, an abnormally high number of Rhizopus infections in surgery patients, with no apparent epidemiologic links, were reported in Argentina. To determine the likelihood of a common source of the cluster, we performed whole-genome sequencing on samples collected during 2006–2014. Most isolates were separated by >60 single-nucleotide polymorphisms, and we found no evidence for recombination or nonneutral mutation accumulation; these findings do not support common source or patient-to-patient transmission. Assembled genomes of most isolates were ≈25 Mbp, and multiple isolates had substantially larger assembled genomes (43–51 Mbp), indicative of infections with strain types that underwent genome expansion. Whole-genome sequencing has become an essential tool for studying epidemiology of fungal infections. Less discriminatory techniques may miss true relationships, possibly resulting in inappropriate attribution of point source.
Mucormycosis is a debilitating fungal infection; the mortality rate among persons with predisposing factors such as skin trauma (e.g., surgery), diabetes mellitus, or organ transplant is high. The fungus can be directly inoculated into a wound or inhaled (1–3). Rhizopus spp. are the Mucorales fungi that most commonly cause mucormycosis (1,2,4) and are the most common non-Aspergillus cause of invasive filamentous fungal infections (5). However, although Mucorales fungi are ubiquitous in the environment, mucormycosis is relatively uncommon.
Rhizopus microsporus has been shown to be a cause of serious infections after anterior cruciate ligament reconstruction surgeries in Argentina (6,7). A recent review of 40 Rhizopus-associated cases of osteomyelitis that developed after these surgeries from 2005 through 2017 in several regions across Argentina identified 3 species—R. microsporus var. rhizopodiformis, R. microsporus var. microsporus, and R. arrhizus [syn. R. oryzae]—and implicated healthcare practices and facility shortcomings in the infections (8). Limited molecular analyses of osteomyelitis-associated R. microsporus infections identified commonalities among isolated strains (7); however, no genomic epidemiologic analyses have been performed on this nosocomial cluster. In this study, we analyzed the genomes of R. microsporus var. rhizopodiformis isolates from patients from multiple facilities in Argentina in the context of unrelated controls from outside the geographic area to empirically establish the relationships among them and determine whether infections may have originated from a common source.
During 2006–2014, we collected 24 R. microsporus isolates from patients at 14 healthcare facilities in 10 provinces in Argentina (8). For unrelated control isolates, used to establish genomic context for the nosocomial cluster in Argentina, we selected 13 isolates from the US Centers for Disease Control and Prevention (Atlanta, GA, USA), collected from 2003 through 2015 (Table).
We extracted DNA from the 37 isolates by using a DNeasy Blood and Tissue Kit (QIAGEN, https://www.qiagen.com), according to the manufacturer’s recommendations. Genomic DNA was fragmented to ≈500 bp by using a QSonica Q800R2 Sonicator (https://www.sonicator.com), genome libraries were prepared for paired-end sequencing and quantified by using a KAPA Hyper Prep Kit and KAPA Library Quantification Kit (KAPA Biosystems, https://sequencing.roche.com), and 33 samples were sequenced on the Illumina NextSeq at 150 × 150–bp reads and 4 samples on the Illumina MiSeq at 300 × 300–bp reads (both https://www.illumina.com). We deposited Illumina read data in the National Center for Biotechnology Information Sequence read archive (https://www.ncbi.nlm.nih.gov/sra) under BioProject PRJNA526061. We also prepared sample B11533 for long-read sequencing. We extracted high molecular weight DNA with the MasterPure Yeast DNA Purification Kit (Lucigen Epicentre, https://www.lucigen.com) by using a nonenzymatic method for lysis targeting a 20-kb insert size. We performed single-molecule real-time (SMRT) sequencing by using the PacBio RS II SMRT DNA sequencing system (Pacific Biosciences, https://www.pacb.com) as previously described (9). Specifically, we generated 20-kb libraries with the SMRTbell template prep kit 1.0 (Pacific Biosciences). We bound libraries to polymerase by using a DNA/Polymerase Binding Kit P6 v2 (Pacific Biosciences), loaded on 2 SMRT cells (Pacific Biosciences), and sequenced with C4 v2 chemistry (Pacific Biosciences) for 360-min movies. We downloaded the public reference genomes of comparable size to the samples in this study—GCA_002083735 (ATCC 11559), GCA_002708625 (ATCC 52813), and GCA_002083745 (ATCC 52814)—and used them for comparison.
We assembled short read data by using UGAP (https://github.com/jasonsahl/UGAP), which uses the SPAdes genome assembler (10), and assembled the PacBio long reads of sample B11533 by using Canu (11); we performed error correction by using the Illumina short reads in 6 rounds of Pilon (12). Whole-genome single-nucleotide polymorphism (SNP) typing (WGST) included only the 32 genomes (of 37 total) that assembled to ≈25 Mbp. For WGST, we generated SNP matrices to identify point mutations among the isolates (and thus infer strain relatedness) with NASP (13), in which reads were aligned to the assembly of sample B11533 by using the Burrows-Wheeler Alignment tool (14). We called SNPs with the Genome Analysis Toolkit (15) and included them in further analyses only if they were present in all samples, covered by >10× depth with >90% consensus in each sample and not in any duplicated regions in the reference genome as identified by NUCmer (16). The resulting SNP matrix comprised the core genome common to all samples in the analysis. We performed maximum-likelihood phylogenetic analyses with IQ-TREE (17) and maximum-parsimony analyses with MEGA version 7.0 (18), and we constructed phylogenetic trees in iTOL version 3 (19).
We assessed the spatial distribution of SNPs among the Rhizopus genomes by using RecomboMamba, which is part of the RECAP toolbox (https://github.com/TGenNorth/RECAP). RecomboMamba was designed to easily detect regions of relatively high SNP density that may indicate recombination or regions under selection that may confound phylogenetic inference. It uses output from an SNP analysis pipeline and a sliding window to tally the numbers of SNPs for each sample by reference genome position to build a graphic display of SNP density, read depth, and pairwise homoplasy index (20).
Read lengths from the PacBio sequencing of B11533 averaged 2,175 bp. The assembly of PacBio and Illumina data of this genome resulted in a genome size of 27.7 Mbp. Approximately 22.6% of the genome was identified as repeat regions according to NUCmer (16) in the NASP analysis, which is consistent with the size and repeat region variation characteristic of Rhizopus (4). We uploaded this assembly into GenBank (accession no. SMRR00000000).
Genomic Relationships among Isolates
Figure
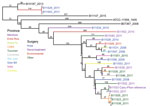
Figure. Maximum parsimony phylogenetic analysis, including 100 bootstrap statistical replicates, and epidemiologic data for Rhizopus microsporus var. rhizopodiformis, including 21 isolates from Argentina, 2006–2014; 3 control isolates (in black); and the public...
Using the whole-gene sequencing (WGS) data, we confirmed that most (22 of 24) of the isolates from the Argentina cluster were R. microsporus var. rhizopodiformis by WGST and by 18S, internal transcribed spacer, 28S, and act1 genetic typing (21,22). A total of 21 isolates fell into a single clade that also included 3 controls and the publicly available genome of the American Type Culture Collection (ATCC) 11559 strain, first described in 1935 in the USSR (23), with 3,170 SNPs among them (Figure). The isolates were collected from patients who had undergone various types of surgeries, encompassing a wide geographic range across multiple years (Figure), and from patients of various ages (Table). Most of the Argentina cluster isolates (n = 17) formed a well-supported inner clade consisting of 1,235 SNPs (Figure). Although 2 sets of epidemiologically related isolates were separated by <20 SNPs, the closest relationship between any other 2 isolates in the tree was 60 SNPs (range 60–912, mean 430), a considerable evolutionary distance, not indicative of a recent transference. A set of 3 samples from the same patient (B11523, B11538, and B11555) were appropriately closely related; the first 2 isolates were identical (i.e., 0 SNPs) and the third was separated by 17 SNPs. One pair of isolates outside the large cluster clade (B11529 and B11543) were separated by 6 SNPs and were collected from 2 patients from the same facility, whose surgeries were 3 weeks apart. This low number of SNPs is characteristic of recent direct transmission or indirect transmission from a common source. The SNP-based phylogenetic analysis included 21.3 Mbp, which covers 99% of the 21.5 Mbp of the unduplicated reference genome of B11533, derived from the 27.7-Mbp full assembly minus the 22.6% of the genome identified as repeat regions. The 21.3-Mbp finding indicates that insertions/deletions may have played a small, if any, role in the evolutionary history of this sample set.
We found no apparent evidence of recombination or selective force in the SNP distribution that could potentially skew phylogenetic inference across the 25 genomes (Appendix Figure 1). The even distribution of SNPs is characteristic of neutral point mutations.
Genome Variability
Five isolates had assembled genome sizes of 43 to 51 Mbp; all other assemblies were ≈25 Mbp (Table). The 5 isolates included 3 from the Argentina cluster, of which 2 were typed as R. microsporus var. microsporus and 1 as R. microsporus var. rhizopodiformis, and 2 controls, which were typed as R. microsporus var. oligosporus by 18S, 28S, ITS, act1, EF-1α sequences (21,22). NASP analysis showed that these samples had multiple SNP states, indicating heterozygosity at several of the SNP loci identified by NASP. Heterozygosity may result from genome expansion and aneuploidy or sample mixtures. Including these 5 samples in the phylogenetic analyses made results inconclusive; therefore, we removed them from the analyses. Sequence data from 3 control samples (all R. microsporus microsporus isolated from skin, respiratory tract, and an unknown source) also contained sequences from Burkholderia rhizoxinica (Table), a known endosymbiont of some R. microsporus strains (24).
An analysis of all samples with ≈25-Mbp genomes (which included 11 control isolates, 3 genomes from public databases, and 21 Argentina cluster isolates) illustrated large genomic distances among different isolates of R. microsporus, even within a variety (Appendix Figure 2). SNP analysis identified 1.2 million SNPs, compared with 3,170 SNPs identified within the cluster. The phylogeny shows that the public genomes for ATCC 52813 and ATCC 52814 differ from the closest control isolates by >800k SNPs, despite all having been identified as R. microsporus var. microsporus, which is remarkable considering that R. microsporus var. rhizopodiformis differs from R. microsporus var. microsporus control isolates by >500k SNPs. Overall, the R. microsporus var. rhizopodiformis group is a relatively tight genomic cluster compared with R. microsporus var. microsporus, possibly because of sampling bias or differential rates of evolution (Appendix Figure 2).
The genomic diversity among isolates from the Argentina cluster of R. microsporus infections is not consistent with a point-source outbreak (25–27). We identified no associations between isolate phylogenetic placement and patient metadata, which included facility, procedure type, and province. Given the extent of genomic differences among the isolates from the cluster and the lack of associations between genotypes and epidemiologic factors, we found no evidence to support the hypotheses of patient-to-patient transmission or a common source. However, our data do not rule out the possibility of a common source because different strains or even species may come from a common source (28). In our case series, the most likely source of infection was environmental contamination at the facilities or during hospital practices (8); contamination of the operating room with unfiltered ambient air might be the common source. During our previous epidemiologic investigation (8), the only common factor among the patients studied was the use of an operating room (for arthroscopy of the knee for anterior cruciate ligament repair, placement of an implantable central venous catheter, or organ transplantation). This speculation is supported by findings from our previous epidemiologic investigation: operating rooms used for case-patients had no HEPA filters; if used, HEPA filters were not used properly; or the operating room was contaminated with unfiltered external air (8).
WGST has become an essential tool for investigating outbreaks of fungal infections; however, defining levels of SNP identity among isolates to determine relatedness remains challenging. Recent WGST analyses of several fungal infection outbreaks help shape our understanding about the relatedness of isolates from point-source outbreaks (25–27,29,30). However, WGS data from clusters not linked to a common source are scarce, and information about the expected genomic diversity among strains from the same region that cannot be linked to a common source is lacking. On the basis of WGS from outbreaks with strong epidemiologic data implicating a common source, isolates that differ by <10 SNPs are considered to be nearly identical and to originate from the same source; isolates sharing tens or hundreds of SNPs are considered different. However, these thresholds are arbitrary, dependent on bioinformatics pipelines, and species specific. To address this issue, Chow et al. defined pairwise SNP distances among isolates of Candida auris from the same patient as an identity reference point (31). Specifically, outbreak isolates are considered to be of the same origin if the number of SNPs between them is the same or lower than the average number of SNPs between multiple isolates from the same patient or known source. Although developed specifically to determine transmission of C. auris, this approach can be adapted to other species and outbreak situations if multiple isolates from the same patients are collected. The number of SNPs separating genomes in the inner clade of the phylogeny generated in this study, which included most of the Argentina isolates from the cluster, was relatively low compared with the number of SNPs separating genomes of the control isolates. However, this number was higher than the differences among multiple isolates from a single patient. Specifically, 0–17 SNPs separated isolates from the same patient, and 60–762 SNPs separated strains from different patients and different facilities. One case of apparent nosocomial transmission was identified in which 2 isolates from 2 patients admitted to the same hospital within 3 weeks differed by 6 SNPs. Because Rhizopus spp. infection is not contagious, transmission probably occurred through the contaminated equipment or from the same environmental source.
Because fungal genomes are large and highly complex, thousands of SNPs separating conspecifics is not uncommon (25,26,29), which is illustrated here within the R. microsporus var. microsporus group. The relatively low numbers of SNPs separating genomes in the inner clade is consistent with a common geographic origin and suggest a relatively recent common ancestor for these 17 isolates. Such limited population diversity is similar to that found for recently emerged fungal populations that display years to decades of evolution in a restricted geographic locale, such as the emergent clones of Cryptococcus gattii in the Pacific Northwest (32) and the recently described clonal population of Coccidioides immitis in southeastern Washington state (33).
Although the mutation rate within R. microsporus is not known, we found no association between genetic distance and sampling dates by using root-to-tip regression analysis, which suggests a lack of molecular clock–like behavior. Furthermore, we found no apparent evidence of recombination or mutation selection in the even SNP distribution across the 25 genomes, suggesting that most SNPs resulted from neutral point mutations and showing that these samples are separated by substantial amounts of evolution, which is not typical of patient-to-patient or point-source outbreaks. However, we cannot rule out the possibility of rapid mutations occurring within a common-source outbreak or a well-established but minimally diverse common-source population. The recent global expansion of C. auris has advanced our knowledge of the varying evolutionary rate of nosocomial fungi; a recent analysis established a within-hospital rate of 5.7 × 10–5 nt substitutions/site/year (34). However, whether the mutation rate of C. auris is applicable to that of Rhizopus spp., a different taxonomic group with different ecology, remains unclear. Such a rate in R. microsporus would predict >1,425 SNPs between genomes separated by only a year. The inclusion of the ATCC 11559 control strain, isolated in 1935 in the Soviet Union, indicates that hypermutation is not occurring because this 84-year-old strain is separated from the Argentina cluster clade by <800 SNPs.
Rhizopus spp. are known to undergo chromosomal duplication events and potentially cross-species hybridization and to contain large proportions of inactive transposable elements (4), which may explain the vast differences in genome sizes and multiple SNP states (i.e., heterozygosity) detected at many genomic loci in the samples with 43–51 Mbp assembled genome sizes. The public genomes for R. microsporus in GenBank are also of various assembly sizes, ranging from 24.1 Mbp (GenBank accession no. GCA_002083735) to 75.1 Mbp (GCA_000697275). A substantial expanse of genome size variation could also result from suboptimal sequence data quality or read length, preventing proper contig formation during assembly and overestimation of genome size or pileup of repeat regions, thereby leading to underestimation of genome size (4). However, our data were of high quality and SNPs were filtered for high-confidence SNPs, although strain mixtures cannot be ruled out. In addition, hybridization between Rhizopus species or subspecies varieties has been described (4), which would confound phylogenetic analysis. Last, many fungi carry bacterial endosymbionts that alter the assembled genome sizes and GC content, including some strains of R. microsporus and Burkholderia rhizoxinicus (35); B. rhizoxinicus often requires obligate symbiosis with R. microsporus (36) and provides a toxin for plant pathogenesis to its host (36,37). In our sample set, 3 control isolates of R. microsporus var. microsporus from skin, respiratory tract, and an unknown source harbored B. rhizoxinicus. To our knowledge, whether the toxin or another factor from the symbiosis contributes to human infection has not been studied. Because many fungi are capable of these and other forms of genomic and chromosomal plasticity, phylogenetic analyses of fungal clusters, even in outbreak scenarios, need to account for these potentially confounding factors.
The cryptic diversity seen in this study might be missed by use of less discriminatory typing techniques, such as matrix-assisted laser desorption/ionization time-of-flight mass spectrometry or repetitive element palindromic PCR (7), possibly resulting in inappropriate point-source attribution. WGST has become the standard for molecular/genomic epidemiology, even (or especially) with understudied or rare pathogen events. However, despite the successful use of WGST to solve numerous medical and public health mysteries, the complexities of certain microbes and their resultant patient clusters are not always clarified, and without intensive sampling and routine genomic surveillance, causes of such clusters may remain hidden.
Dr. Bowers is a research assistant professor at the Translational Genomics Research Institute, Pathogen and Microbiome Division. Her research interests are pathogen tracking and outbreak analyses.
Acknowledgments
We acknowledge Daniel Stamboulian and Joyce G. Peterson for help with culturing the isolates. We also acknowledge Dhwani Batra and Lori A. Rowe for PacBio sequencing.
Funding for this study was provided by Centers for Disease Control and Prevention contract 200-2016-92313, under the Advanced Molecular Detection Initiative.
References
- Jeong W, Keighley C, Wolfe R, Lee WL, Slavin MA, Kong DCM, et al. The epidemiology and clinical manifestations of mucormycosis: a systematic review and meta-analysis of case reports. Clin Microbiol Infect. 2018.PubMedGoogle Scholar
- Petrikkos G, Skiada A, Lortholary O, Roilides E, Walsh TJ, Kontoyiannis DP. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54(Suppl 1):S23–34. DOIPubMedGoogle Scholar
- Neblett Fanfair R, Benedict K, Bos J, Bennett SD, Lo YC, Adebanjo T, et al. Necrotizing cutaneous mucormycosis after a tornado in Joplin, Missouri, in 2011. N Engl J Med. 2012;367:2214–25. DOIPubMedGoogle Scholar
- Gryganskyi AP, Golan J, Dolatabadi S, Mondo S, Robb S, Idnurm A, et al. Phylogenetic and phylogenomic definition of Rhizopus species. G3 (Bethesda). 2018;8:2007–18. DOIPubMedGoogle Scholar
- Slavin M, van Hal S, Sorrell TC, Lee A, Marriott DJ, Daveson K, et al. Invasive infections due to filamentous fungi other than Aspergillus: epidemiology and determinants of mortality. Clin Microbiol Infect. 2015;21:490 e1–10.
- Muscolo DL, Carbo L, Aponte-Tinao LA, Ayerza MA, Makino A. Massive bone loss from fungal infection after anterior cruciate ligament arthroscopic reconstruction. Clin Orthop Relat Res. 2009;467:2420–5. DOIPubMedGoogle Scholar
- Gamarra S, Chaves MS, Cabeza MS, Macedo D, Leonardelli F, Franco D, et al. Mucormycosis outbreak due to Rhizopus microsporus after arthroscopic anterior cruciate ligament reconstruction surgery evaluated by RAPD and MALDI-TOF Mass spectrometry. J Mycol Med. 2018;28:617–22. DOIPubMedGoogle Scholar
- Dignani MC, Santander J, Refojo N, Abrantes R, Hevia A, Clara L, et al. Osteomielitis por mucorales (OMM) asociada a artroscopía de reparación de ligamento cruzado anterior: investigación epidemiológica y recomendaciones para su prevención. Artroscopia. 2018;25:92–9.
- Chow NA, Gade L, Batra D, Rowe LA, Juieng P, Ben-Ami R, et al. Genome sequence of a multidrug-resistant Candida haemulonii isolate from a patient with chronic leg ulcers in Israel. Genome Announc. 2018;6:e00176–18. DOIPubMedGoogle Scholar
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. DOIPubMedGoogle Scholar
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36. DOIPubMedGoogle Scholar
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:
e112963 . DOIPubMedGoogle Scholar - Sahl JW, Lemmer D, Travis J, Schupp JM, Gillece JD, Aziz M, et al. NASP: an accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb Genom. 2016;2:
e000074 . DOIPubMedGoogle Scholar - Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. DOIPubMedGoogle Scholar
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. DOIPubMedGoogle Scholar
- Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–83. DOIPubMedGoogle Scholar
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. DOIPubMedGoogle Scholar
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. DOIPubMedGoogle Scholar
- Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39(suppl):
W475-8 . DOIPubMedGoogle Scholar - Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172:2665–81. DOIPubMedGoogle Scholar
- Abe A, Oda Y, Asano K, Sone T. The molecular phylogeny of the genus Rhizopus based on rDNA sequences. Biosci Biotechnol Biochem. 2006;70:2387–93. DOIPubMedGoogle Scholar
- Abe A, Asano K, Sone T. A molecular phylogeny-based taxonomy of the genus Rhizopus. Biosci Biotechnol Biochem. 2010;74:1325–31. DOIPubMedGoogle Scholar
- Liou GY, Chen SR, Wei YH, Lee FL, Fu HM, Yuan GF, et al. Polyphasic approach to the taxonomy of the Rhizopus stolonifer group. Mycol Res. 2007;111:196–203. DOIPubMedGoogle Scholar
- Partida-Martinez LP, Hertweck C. Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature. 2005;437:884–8. DOIPubMedGoogle Scholar
- Engelthaler DM, Chiller T, Schupp JA, Colvin J, Beckstrom-Sternberg SM, Driebe EM, et al. Next-generation sequencing of Coccidioides immitis isolated during cluster investigation. Emerg Infect Dis. 2011;17:227–32. DOIPubMedGoogle Scholar
- Etienne KA, Roe CC, Smith RM, Vallabhaneni S, Duarte C, Escadon P, et al. Whole-genome sequencing to determine origin of multinational outbreak of Sarocladium kiliense bloodstream infections. Emerg Infect Dis. 2016;22:476–81. DOIPubMedGoogle Scholar
- Litvintseva AP, Hurst S, Gade L, Frace MA, Hilsabeck R, Schupp JM, et al. Whole-genome analysis of Exserohilum rostratum from an outbreak of fungal meningitis and other infections. J Clin Microbiol. 2014;52:3216–22. DOIPubMedGoogle Scholar
- Lockhart SR, Pham CD, Gade L, Iqbal N, Scheel CM, Cleveland AA, et al. Preliminary laboratory report of fungal infections associated with contaminated methylprednisolone injections. J Clin Microbiol. 2013;51:2654–61. DOIPubMedGoogle Scholar
- Etienne KA, Gillece J, Hilsabeck R, Schupp JM, Colman R, Lockhart SR, et al. Whole genome sequence typing to investigate the Apophysomyces outbreak following a tornado in Joplin, Missouri, 2011. PLoS One. 2012;7:
e49989 . DOIPubMedGoogle Scholar - Vaux S, Criscuolo A, Desnos-Ollivier M, Diancourt L, Tarnaud C, Vandenbogaert M, et al.; Geotrichum Investigation Group. Multicenter outbreak of infections by Saprochaete clavata, an unrecognized opportunistic fungal pathogen. MBio. 2014;5:e02309–14. DOIPubMedGoogle Scholar
- Chow NA, Gade L, Tsay SV, Forsberg K, Greenko JA, Southwick KL, et al.; US Candida auris Investigation Team. Multiple introductions and subsequent transmission of multidrug-resistant Candida auris in the USA: a molecular epidemiological survey. Lancet Infect Dis. 2018;18:1377–84. DOIPubMedGoogle Scholar
- Gillece JD, Schupp JM, Balajee SA, Harris J, Pearson T, Yan Y, et al. Whole genome sequence analysis of Cryptococcus gattii from the Pacific Northwest reveals unexpected diversity. PLoS One. 2011;6:
e28550 . DOIPubMedGoogle Scholar - Oltean HN, Etienne KA, Roe CC, Gade L, McCotter OZ, Engelthaler DM, et al. Utility of whole-genome sequencing to ascertain locally acquired cases of coccidioidomycosis in Washington State. Emerg Infect Dis. 2019;25:501–6. DOIPubMedGoogle Scholar
- Rhodes J, Abdolrasouli A, Farrer RA, Cuomo CA, Aanensen DM, Armstrong-James D, et al. Author Correction: Genomic epidemiology of the UK outbreak of the emerging human fungal pathogen Candida auris. Emerg Microbes Infect. 2018;7:104. DOIPubMedGoogle Scholar
- Horn F, Üzüm Z, Möbius N, Guthke R, Linde J, Hertweck C. Draft genome sequences of symbiotic and nonsymbiotic Rhizopus microsporus strains CBS 344.29 and ATCC 62417. Genome Announc. 2015;3:e01370–14. DOIPubMedGoogle Scholar
- Partida-Martinez LP, Monajembashi S, Greulich KO, Hertweck C. Endosymbiont-dependent host reproduction maintains bacterial-fungal mutualism. Curr Biol. 2007;17:773–7. DOIPubMedGoogle Scholar
- Lastovetsky OA, Gaspar ML, Mondo SJ, LaButti KM, Sandor L, Grigoriev IV, et al. Lipid metabolic changes in an early divergent fungus govern the establishment of a mutualistic symbiosis with endobacteria. Proc Natl Acad Sci U S A. 2016;113:15102–7. DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleOriginal Publication Date: April 06, 2020
Table of Contents – Volume 26, Number 5—May 2020
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Address for correspondence:Jolene Bowers, Translational Genomics Research Institute–TGen North, 3051 W. Shamrell Blvd, Flagstaff, AZ 86001, USA
Top