Volume 26, Number 7—July 2020
Research
Human Adenovirus Type 55 Distribution, Regional Persistence, and Genetic Variability
Jun Hang , Adriana E. Kajon, Paul C. F. Graf1, Irina Maljkovic Berry, Yu Yang, Mark A. Sanborn, Christian K. Fung, Anima Adhikari, Melinda S. Balansay-Ames, Christopher A. Myers, Leonard N. Binn, Richard G. Jarman, Robert A. Kuschner, and Natalie D. Collins
, Adriana E. Kajon, Paul C. F. Graf1, Irina Maljkovic Berry, Yu Yang, Mark A. Sanborn, Christian K. Fung, Anima Adhikari, Melinda S. Balansay-Ames, Christopher A. Myers, Leonard N. Binn, Richard G. Jarman, Robert A. Kuschner, and Natalie D. Collins
Figure 1
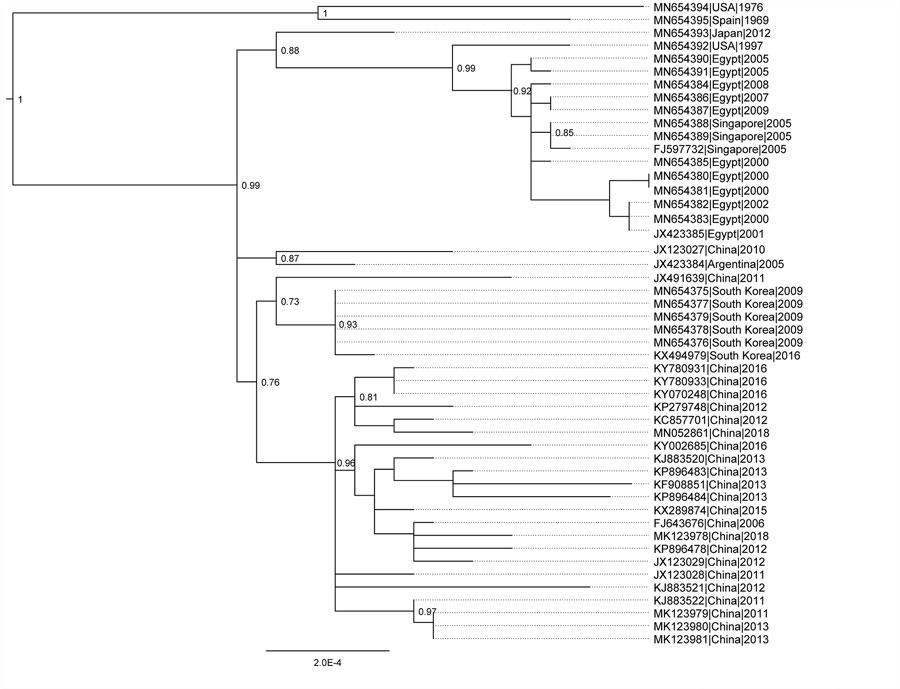
Figure 1. Phylogeny of HAdV-55 based on whole-genome sequences for study of virus distribution, regional persistence, and genetic variability. The phylogenetic tree was generated using the maximum-likelihood method with subtree pruning and regrafting and nearest-neighbor interchange tree search and the Shimodiara-Hasegawa approximate likelihood ratio test for node confidence values. Node confidence values were estimated using approximate likelihood ratio test and the tree was rooted on a HAdV-14 clade as an outgroup (not shown). GenBank accession numbers for isolates are provided. Scale bar indicates node confidence value. HAdV, human adenovirus.
1Current affiliation: US Naval Medical Research Unit 6, Lima, Peru.
Page created: May 06, 2020
Page updated: June 18, 2020
Page reviewed: June 18, 2020
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.