Volume 27, Number 1—January 2021
Research Letter
Relapsing Fever Group Borreliae in Human-Biting Soft Ticks, Brazil
Figure
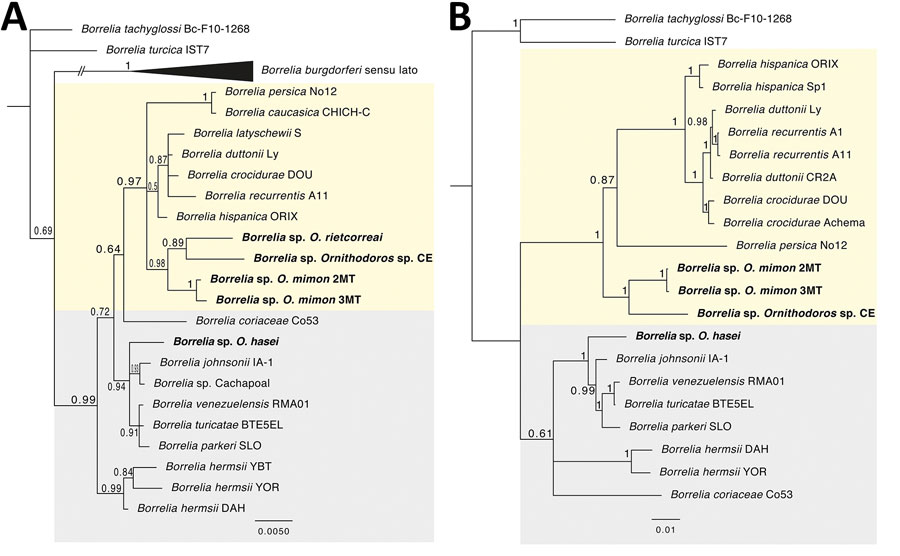
Figure. Bayesian phylogenetic trees inferred for the Borrelia spp. characterized in study of relapsing fever group borreliae in human-biting soft ticks, Brazil. A) Ambiguous alignments of single 16S rRNA gene (1,274 bp); B) concatenated 16S rRNA-flaB-glpQ genes (2,435 bp). Bold indicates borreliae from this study. Trees are drawn to scale. Four independent Markov chain runs for 1,000,000 metropolis-coupled MCMC generations were implemented for the analyses, sampling a tree every 100th generation. The first 25% of the trees represented burn-in, and the remaining trees were used to calculate Bayesian posterior probability values. Both trees were inferred using the Hasegawa-Kishino-Yano model with gamma distribution. Numbers above or below tree branches represent Bayesian posterior probabilities. Light yellow and gray backgrounds denote Old World and New World relapsing fever group Borrelia spp., respectively. Scale bar indicates nucleotide substitutions per site.
1Current affiliation: Universidad de Concepción, Chillán, Ñuble, Chile.