Volume 27, Number 5—May 2021
Research Letter
Novel Mutation of SARS-CoV-2, Vietnam, July 2020
Figure
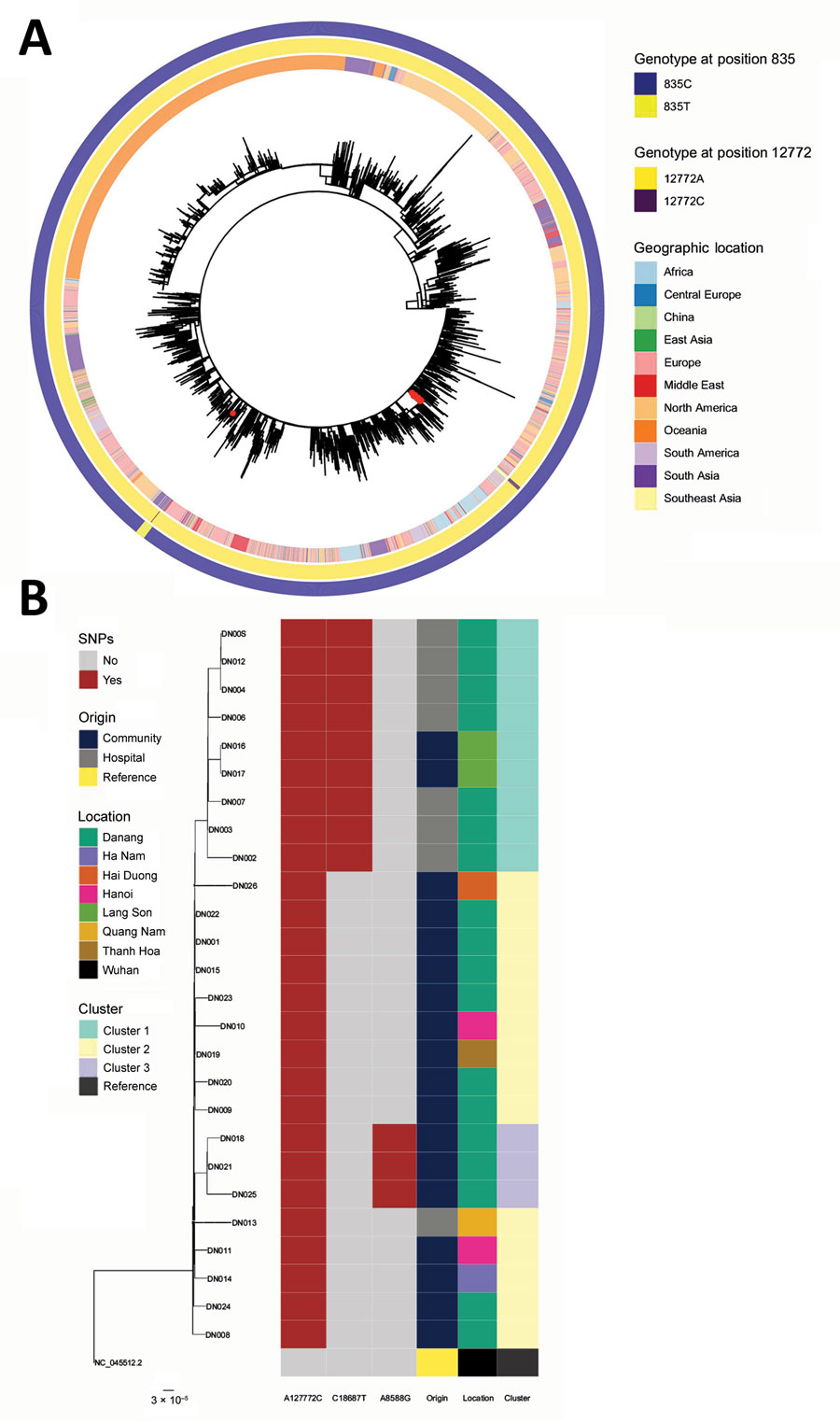
Figure. Maximum likelihood phylogenetic trees of SARS-CoV-2 B.1.1 lineage sequences globally and sequences from Danang, Vietnam. A) Global maximum-likelihood phylogenetic tree of SARS-CoV-2 B.1.1 lineage. The phylogeny was inferred with the general time-reversible plus frequencies model using 1,000 bootstrap replicates. Red dots represent viruses from the Danang cluster. The outer ring shows lineage as determined using Pangolin (https://github.com/cov-lineages/pangolin/releases/tag/v2.3.0), and the inner ring shows the geographic location of collection. B) Maximum-likelihood phylogenetic tree built from 26 Danang-related SARS-CoV-2 sequences (represented by DN plus a 3-digit number); the Wuhan strain genome (GenBank accession no. NC_045512.2) is an outgroup. Columns to the right show the nucleotide variation in 3 locations on the SARS-CoV-2 genome that define phylogenetic clusters in the Danang cluster with their origin, the location the patients were found, and the cluster of the sequence. The ModelFinder Plus option Hasegawa-Kishino-Yano substitution model, including modelling of amino acid frequencies was the best model for these samples. Scale bar indicates substitutions per site. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
1These authors equally contributed to this work.