Volume 27, Number 8—August 2021
Research Letter
Whole-Genome Sequencing of SARS-CoV-2 from Quarantine Hotel Outbreak
Lex E.X. Leong , Julien Soubrier, Mark Turra, Emma Denehy, Luke Walters, Karin Kassahn, Geoff Higgins, Tom Dodd, Robert Hall, Katina D’Onise, Nicola Spurrier, Ivan Bastian, and Chuan K. Lim
, Julien Soubrier, Mark Turra, Emma Denehy, Luke Walters, Karin Kassahn, Geoff Higgins, Tom Dodd, Robert Hall, Katina D’Onise, Nicola Spurrier, Ivan Bastian, and Chuan K. Lim
Figure
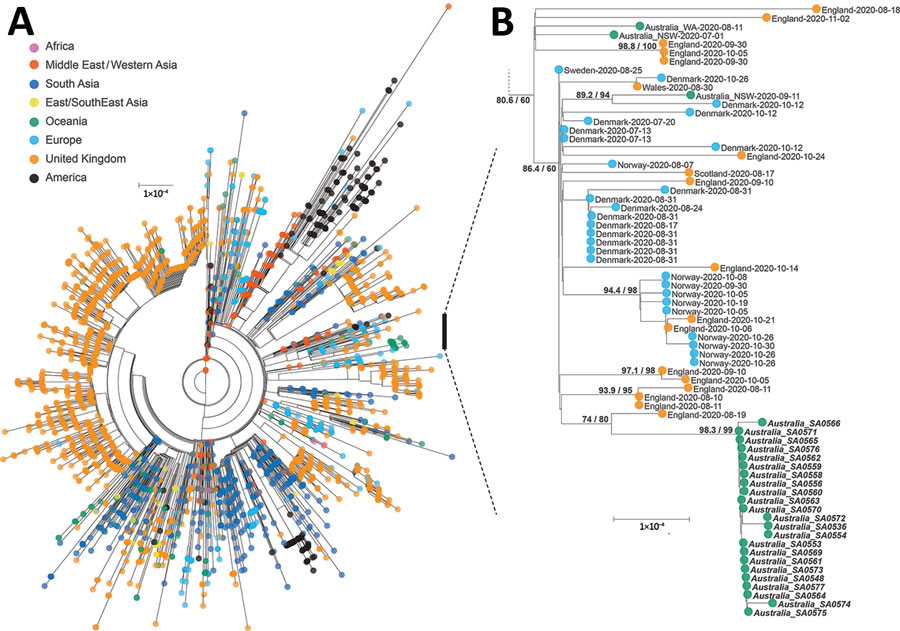
Figure. Maximum-likelihood phylogenetic tree of severe acute respiratory syndrome coronavirus 2 genomes from a quarantine hotel–associated community outbreak of coronavirus disease in South Australia, Australia. A) Genomes from lineage B.1.36 (n = 3,038). B) Subtree of lineage B.1.36.1 focusing on the quarantine hotel clusters and a returned traveler from the United Kingdom; bold type indicates those strains. Consensus genomes were profile-aligned using COVID-Align (5), and phylogenetic trees were constructed using IQ-TREE with general time reversible plus invariate plus gamma 4 sites model (6). SH-like approximate likelihood ratio test score was 98.3%, ultrafast bootstrap approximation 99%. Scale bar indicates substitutions per site.
References
- Miller IF, Becker AD, Grenfell BT, Metcalf CJE. Disease and healthcare burden of COVID-19 in the United States. Nat Med. 2020;26:1212–7. DOIPubMedGoogle Scholar
- Speake H, Phillips A, Chong T, Sikazwe C, Levy A, Lang J, et al. Flight-associated transmission of severe acute respiratory syndrome coronavirus 2 corroborated by whole-genome sequencing. Emerg Infect Dis. 2020;26:2872–80. DOIPubMedGoogle Scholar
- Zhou F, Li J, Lu M, Ma L, Pan Y, Liu X, et al. Tracing asymptomatic SARS-CoV-2 carriers among 3674 hospital staff:a cross-sectional survey. EClinicalMedicine. 2020;26:
100510 . DOIPubMedGoogle Scholar - Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403–7. DOIPubMedGoogle Scholar
- Lemoine F, Blassel L, Voznica J, Gascuel O. COVID-Align: accurate online alignment of hCoV-19 genomes using a profile HMM. Bioinform. 2020;btaa871.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. DOIPubMedGoogle Scholar
- Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Chall. 2017;1:33–46. DOIPubMedGoogle Scholar
- Endo A, Abbott S, Kucharski AJ, Funk S; Centre for the Mathematical Modelling of Infectious Diseases COVID-19 Working Group. Estimating the overdispersion in COVID-19 transmission using outbreak sizes outside China. Wellcome Open Res. 2020;5:67. DOIPubMedGoogle Scholar
- van Dorp L, Richard D, Tan CCS, Shaw LP, Acman M, Balloux F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat Commun. 2020;11:5986. DOIPubMedGoogle Scholar
Page created: June 16, 2021
Page updated: July 19, 2021
Page reviewed: July 19, 2021
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.