Volume 28, Number 6—June 2022
Dispatch
New Variant of Vibrio parahaemolyticus, Sequence Type 3, Serotype O10:K4, China, 2020
Figure 2
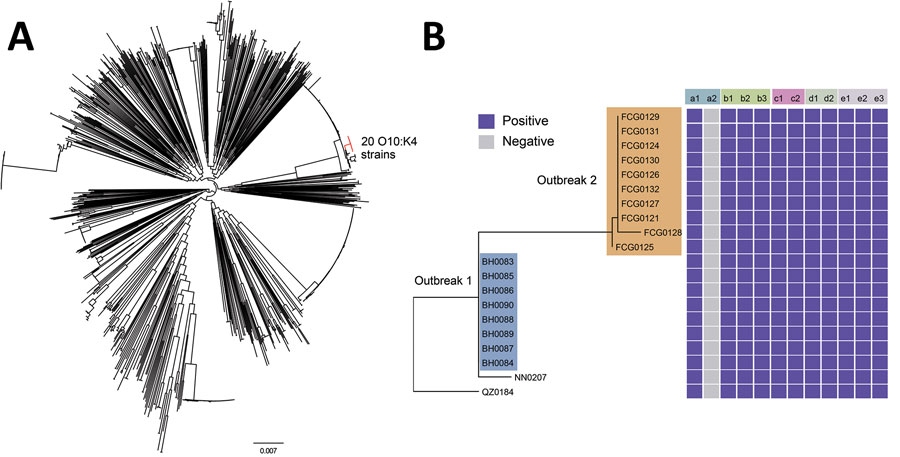
Figure 2. Phylogenetic tree based on the single-nucleotide variations in the core genomes of 1,120 Vibrio parahaemolyticus genomes: 20 isolates from patients in the Beibu Gulf area of Guangxi, China, 33 isolates collected in Guangxi in recent years, and all 1,067 genomic sequences available in the PubMLST database (https://pubmlst.org/organisms/vibrio-parahaemolyticus) (Appendix). A) Maximum-likelihood tree based on the single-nucleotide variations in the nonrepetitive, nonrecombinant regions of the genomes. Branches in red indicate the O10:K4 serotype strains. Scale bar indicates frequency of single-nucleotide variations. B) Distribution of virulence genes, pathogenic islands, secretion systems, characteristic genes in pandemic clones, and antimicrobial resistance genes. a1, tdh; a2, trh; b1, VPaI-2; b2, VPaI-3; b3, VPaI-4; c1, T3SS; c2, T6SS1; d1, orf8; d2, toxRS/new; e1, tet(34); e2, tet(35); e3, blaCARB-22.
1These first authors contributed equally to this article.
2These senior authors contributed equally to this article.