Volume 28, Number 6—June 2022
Research Letter
Multistate Outbreak of Infection with SARS-CoV-2 Omicron Variant after Event in Chicago, Illinois, USA, 2021
Figure
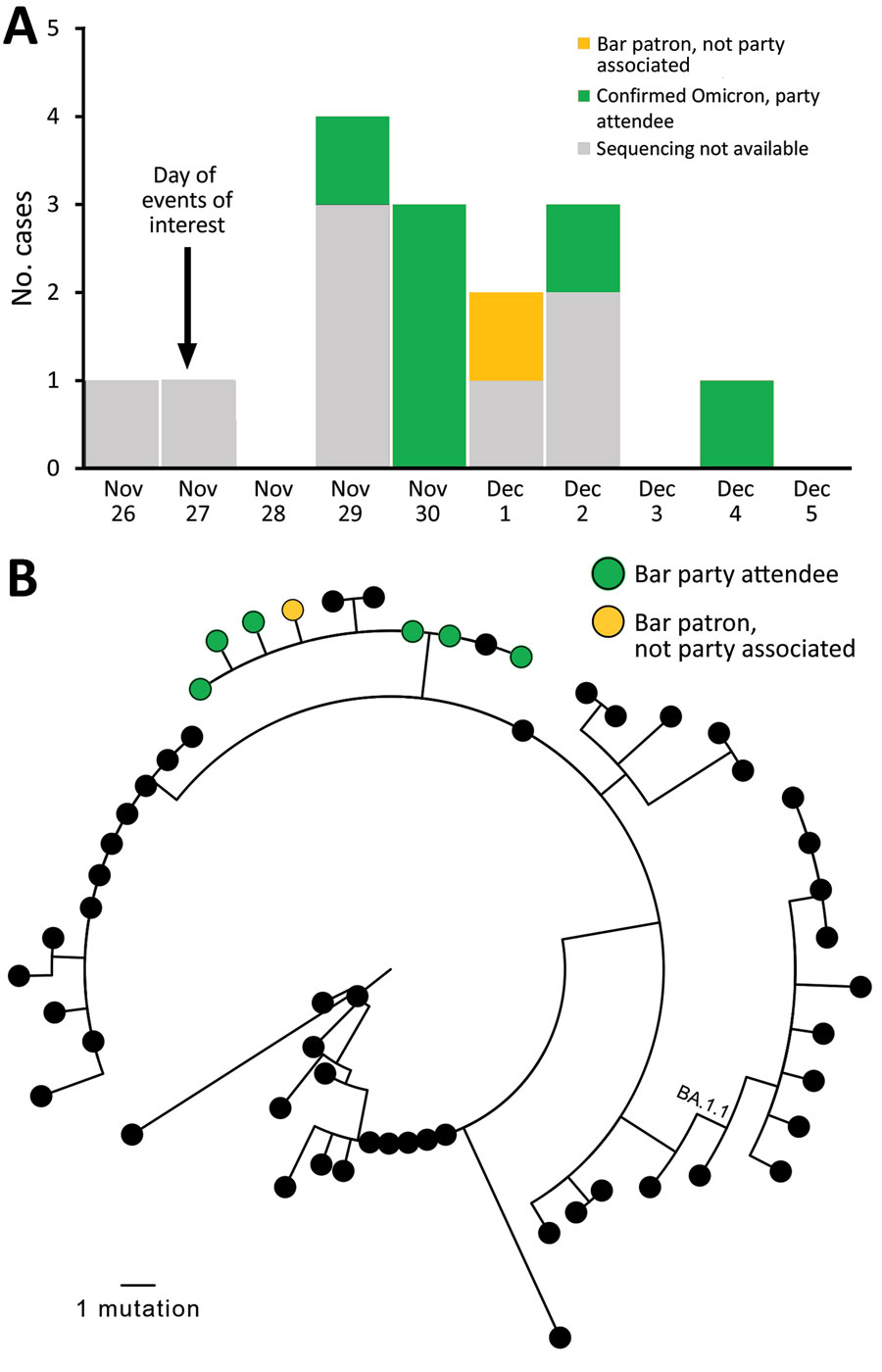
Figure. Cases in multistate (Colorado, Illinois, Louisiana, Missouri, Michigan) outbreak of infection with severe acute respiratory syndrome 2 Omicron variant after event in Chicago, Illinois, USA, November–December 2021. A) Cases over time. Sequencing results are shown, if available. B) Genetic relatedness of viruses isolated. Maximum-likelihood phylogeny of 7 sequenced Omicron samples in bar-associated outbreak (green and yellow) with 50 contextual sequences (black). Contextual sequences are a random sample of Omicron BA.1 and BA.1.1 sequences selected from all Omicron sequences in GISAID (https://www.gisaid.org) that were collected in the United States or before December 11, 2021, and had >90% genome coverage. Random selection was performed by using CLC Genomics Workbench (QIAGEN, https://www.qiagen.com). No contextual sequences were from Illinois. GISAID accession numbers for all included sequences are listed in the Appendix. One outbreak-associated specimen was sequenced by a private laboratory and not uploaded to GISAID. Full-genome sequences were used for PhyML phylogenetic analysis (4), excluding 250 bp from genome ends and an error-prone region (reference positions 21492–21935). Outbreak sequences were identical to each other or contained a single-nucleotide substitution (T12000C, T22813G, T25414C) and clustered (with 3 contextual sequences) in a clade diverged by 2 nts from the closest other sequences. The 2 nt substitutions that defined the outbreak branch (C11950T, C28472T) were present in just 5.2% of contemporaneous Omicron sequences from the United States available on GISAID, indicating that all available outbreak sequences were closely genetically related.
References
- Fisher KA, Tenforde MW, Feldstein LR, Lindsell CJ, Shapiro NI, Files DC, et al.; IVY Network Investigators; CDC COVID-19 Response Team. CDC COVID-19 Response Team. Community and close contact exposures associated with COVID-19 among symptomatic adults ≥18 years in 11 outpatient health care facilities—United States, July 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1258–64. DOIPubMedGoogle Scholar
- Sami S, Turbyfill CR, Daniel-Wayman S, Shonkwiler S, Fisher KA, Kuhring M, et al. Community transmission of SARS-CoV-2 associated with a local bar opening event—Illinois, February 2021. MMWR Morb Mortal Wkly Rep. 2021;70:528–32. DOIPubMedGoogle Scholar
- Majra D, Benson J, Pitts J, Stebbing J. SARS-CoV-2 (COVID-19) superspreader events. J Infect. 2021;82:36–40. DOIPubMedGoogle Scholar
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. DOIPubMedGoogle Scholar
1These authors are co-senior authors.