Volume 29, Number 1—January 2023
Research
Genomic Confirmation of Borrelia garinii, United States
Cite This Article
Citation for Media
Abstract
Lyme disease is a multisystem disorder primarily caused by Borrelia burgdorferi sensu lato. However, B. garinii, which has been identified on islands off the coast of Newfoundland and Labrador, Canada, is a cause of Lyme disease in Eurasia. We report isolation and whole-genome nucleotide sequencing of a B. garinii isolate from a cotton mouse (Peromyscus gossypinus) in South Carolina, USA. We identified a second B. garinii isolate from the same repository. Phylogenetic analysis does not associate these isolates with the previously described isolates of B. garinii from Canada.
Lyme disease is a multisystem disorder caused by infection with bacteria of the Borrelia burgdorferi sensu lato species complex. Three members of this complex, B. burgdorferi sensu stricto, B. garinii, and B. afzelii, are responsible for most cases of Lyme disease worldwide (1,2). B. burgdorferi s.l. is the only one of these 3 species that is found widely in North America, although B. garinii has been identified on islands off the coast of Newfoundland and Labrador, Canada (3–5).
We describe the isolation and genome sequencing characterization of a South Carolina B. garinii isolate from a repository of strains from rodent hosts and tick vectors in the southeastern United States that had been identified as B. burgdorferi s.l. A second B. garinii isolate from the same B. burgdorferi s.l. strain repository was identified on the basis of multilocus sequence typing (MLST). Phylogenetic analysis showed that these 2 strains from the southeastern United States were most closely related to a group of B. garinii isolates from Europe but were not derived from strains from Canada, or vice versa (3).
Sources, Cultivation, and Analyses of Borrelia spp.
The 2 Borrelia isolates described were isolated from ear biopsy specimens from a cotton mouse (Peromyscus gossypinus) (SCCH-7) trapped in Charleston County, South Carolina, in 1995 and from an eastern woodrat (Neotoma floridana) (SCGH-19) trapped in Georgetown County, South Carolina, in 1996 (Appendix) (6). We performed Borrelia culture in Barbour-Stoenner-Kelly H medium, DNA purification, and PCR analyses as described (7). We detected B. burgdorferi s.l. in samples by amplification of the 5S-23S intergenic region (8) (Appendix) by using species-specific PCR and primers designed on the basis of the ospA gene, which confirmed the presence of multiple spirochete species (9). Cultures in which B. garinii was confirmed were plated on solid medium, and clonal single colonies were isolated according to a modified protocol (10) (Appendix).
Whole-Genome Sequencing and Genome Assembly
We performed whole-genome sequencing by using the Pacific Biosciences Sequel II system (https://www.pacb.com). We performed genome assembly by using the Genome Assembly tool in PacBio SMRTLink version 10.2 and 150 Mb of the HiFi reads >5 kb (Appendix).
Nucleotide Sequence Accession Numbers
Sequences have been deposited in GenBank. The genome assembly of SCCH-7 has been deposited in GenBank under BioProject PRJNA431102 and BioSample accession no. SAMN26226110 (Appendix). Nucleotide sequences of 8 housekeeping genes (clpA, clpX, nifS, pepX, pyrG, recG, rplB, and uvrA) of SCCH-7 and SCGT-19 have been deposited in GenBank under accession nos. KP795353‒60 (SCCH-7) and KT285873‒80 (SCGT-19). The MLST sequences have also been deposited into the PubMLST database (https://pubmlst.org) under allele numbers assigned to unique loci (SCCH-7, clpX allele no. 272 and uvrA allele no. 278; SCGT-19, clpA allele no. 311 and clpX gene allele no. 273). Unique sequence type (ST) numbers in the PubMLST database are 1049 for SCCH-7 and 1050 for SCGT-19.
Sequence Analysis
We performed MLST analysis of 8 housekeeping genes (clpA, clpX, nifS, pepX, pyrG, recG, rplB, and uvrA) of both isolates (SCCH-7 and SCGT-19) and whole-genome sequencing of strain SCCH-7 on DNA isolated at passage 6 as described (11). The maximum-likelihood phylogeny of B. garinii strains from a concatenated dataset of the 8 housekeeping loci sequences (184 total isolates, 4,791 nt) was inferred in RaxML (https://raxml-ng.vital-it.ch) under the generalized time reversible plus Γ4 model (Appendix). We performed phylogeographic analysis of diffusion on discrete space as implemented in BEAST (12) under the constant-size coalescent tree prior, and symmetric substitution model with Bayesian stochastic search variable selection enforced (Appendix). To compare sequences for the entire chromosome, we aligned the SCCH-7 chromosomal sequence with the 2 published chromosomal sequences of strain 20047 by using NUCMER (13). We derived a phylogenetic tree by using IQTREE (14) with default parameters from an MLST alignment of 34 B. garinii isolates most closely related to the 2 isolates from the United States.
B. garinii from Rodents in South Carolina
The 2 B. garinii isolates we report were cultured from ear biopsy samples of a cotton mouse (Peromyscus gossypinus) (isolate SCCH-7) and an eastern woodrat (Neotoma floridana) (isolate SCGT-19); both were trapped in South Carolina (6). Those cultures were part of a southeastern United States collection of ≈300 Borrelia isolates that were obtained during 1991‒1999 in Missouri, Georgia, Florida, Texas, and South Carolina and housed in the James H. Oliver, Jr., Institute of Arthropodology and Parasitology, Georgia Southern University (Statesboro, Georgia, USA). Multiple Borrelia species in numerous cultures of this collection, often present as co-infections, were reported in earlier investigations, including B. andersonii (15–18), B. burgdorferi s.s. (6,15,19), B. bissettiae (15–18), B. carolinensis (7,20), B. americana (21), and a previously undescribed isolate from Texas, TXW-1 (16–18).
We confirmed B. burgdorferi s.l. in cultures by PCR amplification of total DNA with a 5S-23S rRNA set of primers (8). We identified the B. burgdorferi s.l. species present by cloning the total PCR products into the pCR4-TOPO TA vector and sequencing individual recombinants. We observed sequences with high similarity to B. garinii from 5 cultures. We then plated those cultures on solid medium to obtain single colonies and chose pure clonal cultures of B. garinii SCCH-7 clone 138 and SCGT-19 clone 19 from 2 of the cultures for further study.
Whole-Genome Sequence of B. garinii Isolate SCCH-7
We determined the whole-genome sequence of isolate SCCH-7 by single-molecule real-time PacBio methods (Appendix). Similar to other B. burgdorferi sensu lato genomes (22–24), the SCCH-7 genome contains a linear chromosome and several linear plasmids (lp) and circular (cp) plasmids. SCCH-7 carries lp17, lp28–7, lp32–10, lp36, and lp54 and cp26, cp32–3, and cp32–6 (25). Plasmid SCCH-7 sequences are typical of known B. garinii genomes and are similar to those of B. garinii strain 20047, although strain 20047 carries an lp28–4 plasmid that is lacking in SCCH-7. The SCCH-7 genome is 1,161,212 bp (chromosome 906,106 bp, linear plasmids 168,083 bp, and circular plasmids 87,023 bp). Because the linear chromosome and plasmid sequences include all telomeres, this genome joins B. burgdorferi B31 and B. mayonii MN14–1539 genomes in being truly complete (26–28). The SCCH-7 chromosome differs from that of B. garinii strain 20047 by only 2 single-nucleotide variations (SNVs) and 2 short insertion/deletions from the sequence in accession no. CP028861 and by 8 SNVs and 4 short indels from the sequence in CP018744 (those 2 20047 chromosomal sequences were produced by 2 independent research groups, those of S. Bontemps-Gallo and G. Margos, Bioproject PRJNA224116). The 20047 plasmids were described briefly by Casjens et al. (24). This whole-genome sequence unambiguously demonstrates that SCCH-7 is a B. garinii isolate.
Phylogenetic Analysis
B. garinii isolates from North America have been previously reported on coastal islands in the Atlantic Ocean in eastern Canada (3–5,29). To investigate whether the South Carolina isolates might have originated from these islands in Canada or vice versa, and to clarify the relationship of SCCH-7 and SCGT-19 with other B. garinii isolates, we amplified by using PCR and determined SCGT-19 sequences for the 8 genes (Appendix) previously used in MLST analyses of B. burgdorferi sensu lato isolates. We then extracted those sequences from the SCCH-7 and 20047 whole-genome sequences.
Figure
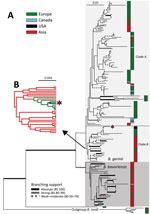
Figure. Maximum-likelihood phylogeny of Borrelia garinii/B. bavariensis. A) Maximum-likelihood phylogeny of B. garinii/B. bavariensis rooted with B. turdi. Topology is based on analysis of the...
We compiled a phylogenetic tree (Appendix Figure 1) of the MLST data from isolates in this branch of the B. burgdorferi s.l. species and a maximum-likelihood (RAxML) tree (Figure) of the MLST sequences that includes isolates SCCH-7 and SCGT-19 and the 178 other isolates available from the closely related species B. garinii and B. bavariensis, as well as 5 isolates of B. turdi as an outgroup (Appendix Table 1). Apart from 1 unusual isolate from European Russia (pubMLST ID:2488 Om16-103-Iapr) that is a sister branch to all the other isolates, the remaining 178 isolates form 2 clades that agree with previously defined B. bavariensis (39 isolates) and B. garinii (139 isolates) species (Figure) (30). The B. bavariensis group contains 4 isolates from Europe and 1 isolate from Canada, interspersed among most isolates from Asia, suggesting, on the basis of maximum parsimony, that this group is an ancestrally clade from Asia that has had several independent introgressions into Europe (31).
The B. garinii clade is split into 2 major clades. The larger one (clade A, 108 isolates) comprises 76 isolates from Europe, 13 from continental Asia, 2 from Japan, 9 from Canada (Newfoundland and Labrador), and 8 from Iceland that are mostly distributed within this branch without apparent clustering by geographic origins. The smaller B. garinii clade (clade B, 31 isolates) contains 21 isolates from continental Asia and Japan, 8 from Europe, and the 2 described here from the United States. The 2 United States isolates form a nested subclade with 5 strains of European origin in clade B (Figure, panel B). Also, the B. garinii from Canada are members of clade A (Figure) and are not closely related to the United States isolates.
To shed more light on the possible origin of the 2 United States isolates and the evolutionary history of B. garinii in general, we performed phylogeographic analysis of diffusion in discrete space as implemented in BEAST (Appendix). This Bayesian method infers the ancestral state at each node of a given discrete trait (in this case, the geographic origin of the strain). The resulting tree topology (Appendix Figure 2, panel A), as inferred under the Coalescence (https://www2.unil.ch/popgen/softwares/quantinemo/coalescence.html) constant size model, separated the isolates into several groups. The B. bavariensis clade and clade B, whose composition corresponds to the MLST topology (Figure), are both predicted to originate in Asia. The 2 United States isolates (Figure) are in clade B and nested among a few sequences from Europe (Appendix Figure 2). The isolates from Europe are split into 3 clades (A1, A2, and A3), the first of which (A1) is found at the base of the whole B. bavariensis/B. garinii portion of the tree. Clade A1 is composed of 5 divergent B. garinii isolates from Slovakia separated from all other isolates from Europe and the B. bavariensis isolates. The ancestral clade from Asia (clade B) (Figure; Appendix Figure 2, panel A) is nested within clade A1 and clade A2, which were both predicted to have origins in Europe. The terminal position of strains from Europe in the ancestral clade from Asia (clade B) suggests a secondary, more recent introduction into Europe from Asia. Thus, ancestral reconstruction with BEAST suggested frequent historical and recent migration events of B. bavarensis and B. garinii species within Eurasia.
The affiliation of United States isolates with Europe and the broader B. garinii ancestral clade from Asia (clade B) is consistently present in both trees (Figure; Appendix Figure 2, panel A) and is supported by phylogeographic reconstruction using the Bayesian stochastic search variable selection algorithm (32–34). However, because of the large single number of isolates and relatively low number of phylogenetic-informative positions (i.e., high sequence similarity), the bootstrap support of inner branches was not high. Therefore, we tested the independent evolutionary history of isolates from the United States and Canada by using the approximately unbiased topology test (35). First, we force-constrained the monophyly of the 2 United States isolates with each of the 9 isolates from Canada; we then used RAxML to reoptimize the general topology. We then compared the per-site log-likelihood scores of those alternative topologies with the original MLST topology (Figure) by using the approximately unbiased in CONSEL (36). The resulting p values, ranging from 1.48 × 10−36 to 1.9 × 10−2 (Appendix Table 2), support the rejection of a common origin of B. garinii from Canada and the United States. We conclude that, in contrast to the isolates from Canada, which might have been introduced there from Europe or Iceland by seabirds and ticks associated with them (37), B. garinii from the southeastern United States are a part of ancestral lineage from East Asia that might have arrived in the United States from Europe.
Chromosomal Relationships with Closely Related B. garinii Genomes
A maximum-likelihood tree of 32 closely related B. garinii genomes based on 8 housekeeping loci (Appendix Figure 3) shows that the 2 isolates from the United States cluster with a few isolates from Europe, which, by tree topology, were probably associated with an ancestor from Asia (Figure). We compiled all sequence differences at the 8 housekeeping loci among the strains that are most closely related to the 2 isolates from the United States (Appendix Table 3). The genome-derived MLST SCCH-7 sequence and 2 independent 20047 MLST sequences are nearly identical. The reported 20047 MSLT sequence has 2 differences in the recG gene compared with the 3 whole-genome sequences, probably caused by sequencing errors. Those sequence identities strongly support a non-Canada origin, specifically a recent Europe origin, of United States isolate SCCH-7.
In contrast to SCCH-7, strain SCGT-19 shows a distinct MLST haplotype defined by 16 SNVs and 1 short indel (Appendix Table 3). The SCGT-19 versions of some of these SNVs and the indel are found in other B. garinii strains from Japan and Europe, suggesting that they are unlikely to be sequencing errors. Furthermore, consecutive runs of SNVs at the clpA and clpX loci strongly indicate that their origins are caused by recombination and not de novo mutation. The differences between SCCH-7 and SCGT-19 suggests that a migration or importation of B. garinii from Eurasia to the United States might have consisted of multiple strains of a source population.
Our results provide strong evidence that B. garinii has been present in rodents in South Carolina, although its current status there is not known. Specifically, 5 samples we tested were positive for B. garinii, and from 2 independent B. garinii cultures, we propagated and analyzed, SCCH-7 clone 138 and SCCH-19 clone 19.
MLST analyses of both isolates and whole-genome sequencing of SCCH-7 showed that these isolates are not closely related to B. garinii strains from Canada; however, they are closely related to a subset of Eurasian isolates. How and when B. garinii arrived in South Carolina remains unknown. There were no reported Lyme disease outbreaks in the southeastern United States in humans at the time the strains were deposited in the repository or during the subsequent 2 decades. This finding minimizes the urgency for an immediate new search for B. garinii in this region. Nonetheless, clinical vigilance for B. garinii in humans in this region seems warranted.
Dr. Rudenko is a senior research scientist at the Institute of Parasitology, Biology Centre Czech Academy of Sciences, Ceske Budejovice, Czech Republic. Her primary research interests include spirochetes from the B. burgdorferi sensu lato complex, and ecologic, epidemiologic, and molecular aspects of Lyme disease.
Acknowledgments
We thank all former members of the James Oliver, Jr. Institute of Arthropodology and Parasitology, Statesboro, GA, for contributing to establishment of the southeastern spirochete collection during 1991–1999, and Xiaohua Yang for her contributions in culturing the microbes.
This study was supported in part by grants AI110820 (C.M.F.) and AI139782 (W.Q.) from the National Institute of Allergy and Infectious Diseases, National Institutes of Health; by grants NV-19-05-00191 from the Ministry of Health of the Czech Republic (N.R and M.G.) and 21-26209S from the Czech Grant Agency (A.H.); the Institute of Parasitology, Biology Centre, Czech Academy of Sciences (N.R., M.G., A.H., and L.G.); the Institute for Genomic Sciences (C.M.F. and E.F.M.), New England BioLabs (R.G.M.); and the Steven and Alexandra Cohen Foundation and the Global Lyme Alliance (B.J.L.).
References
- Radolf JD, Caimano MJ, Stevenson B, Hu LT. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol. 2012;10:87–99. DOIPubMedGoogle Scholar
- Piesman J, Schwan TG. Ecology of borreliae and their arthropod vectors. In: Samuels DS, Radolf JD, editors. Borrelia: molecular biology, host interaction, and pathogenesis. Norfolk (UK): Caister Academic; 2010. p. 251–78.
- Smith RP Jr, Muzaffar SB, Lavers J, Lacombe EH, Cahill BK, Lubelczyk CB, et al. Borrelia garinii in seabird ticks (Ixodes uriae), Atlantic Coast, North America. Emerg Infect Dis. 2006;12:1909–12. DOIPubMedGoogle Scholar
- Munro HJ, Ogden NH, Lindsay LR, Robertson GJ, Whitney H, Lang AS. Evidence for Borrelia bavariensis infections of Ixodes uriae within seabird colonies of the North Atlantic Ocean. Appl Environ Microbiol. 2017;83:e01087–17. DOIPubMedGoogle Scholar
- Baggs EM, Stack SH, Finney-Crawley JR, Simon NP. Peromyscus maniculatus, a possible reservoir host of Borrelia garinii from the Gannet Islands off Newfoundland and Labrador. J Parasitol. 2011;97:792–4. DOIPubMedGoogle Scholar
- Oliver JH Jr, Clark KL, Chandler FW Jr, Tao L, James AM, Banks CW, et al. Isolation, cultivation, and characterization of Borrelia burgdorferi from rodents and ticks in the Charleston area of South Carolina. J Clin Microbiol. 2000;38:120–4. DOIPubMedGoogle Scholar
- Rudenko N, Golovchenko M, Grubhoffer L, Oliver JH Jr. Borrelia carolinensis sp. nov., a new (14th) member of the Borrelia burgdorferi Sensu Lato complex from the southeastern region of the United States. J Clin Microbiol. 2009a;47:134–41. DOIPubMedGoogle Scholar
- Postic D, Assous MV, Grimont PA, Baranton G. Diversity of Borrelia burgdorferi sensu lato evidenced by restriction fragment length polymorphism of rrf (5S)-rrl (23S) intergenic spacer amplicons. Int J Syst Bacteriol. 1994;44:743–52. DOIPubMedGoogle Scholar
- Demaerschalck I, Ben Messaoud A, De Kesel M, Hoyois B, Lobet Y, Hoet P, et al. Simultaneous presence of different Borrelia burgdorferi genospecies in biological fluids of Lyme disease patients. J Clin Microbiol. 1995;33:602–8. DOIPubMedGoogle Scholar
- Rosa PA, Hogan D. Colony formation by Borrelia burgdorferi in solid medium: clonal analysis of osp locus variants. In: Proceedings of the 1st International Conference on Tick-Borne Pathogens at the Host-Vector Interface: An Agenda for Research; 1992 Sep 15–18. St. Paul (MN): University of Minnesota; 1992. p. 95–103.
- Margos G, Gatewood AG, Aanensen DM, Hanincová K, Terekhova D, Vollmer SA, et al. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2008;105:8730–5. DOIPubMedGoogle Scholar
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. DOIPubMedGoogle Scholar
- Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. MUMmer4: A fast and versatile genome alignment system. PLOS Comput Biol. 2018;14:
e1005944 . DOIPubMedGoogle Scholar - Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74. DOIPubMedGoogle Scholar
- Oliver JH Jr, Lin T, Gao L, Clark KL, Banks CW, Durden LA, et al. An enzootic transmission cycle of Lyme borreliosis spirochetes in the southeastern United States. Proc Natl Acad Sci U S A. 2003;100:11642–5. DOIPubMedGoogle Scholar
- Lin T, Oliver JH Jr, Gao L, Kollars TM Jr, Clark KL. Genetic heterogeneity of Borrelia burgdorferi sensu lato in the southern United States based on restriction fragment length polymorphism and sequence analysis. J Clin Microbiol. 2001;39:2500–7. DOIPubMedGoogle Scholar
- Lin T, Oliver JH Jr, Gao L. Genetic diversity of the outer surface protein C gene of southern Borrelia isolates and its possible epidemiological, clinical, and pathogenetic implications. J Clin Microbiol. 2002;40:2572–83. DOIPubMedGoogle Scholar
- Lin T, Oliver JH Jr, Gao L. Comparative analysis of Borrelia isolates from southeastern USA based on randomly amplified polymorphic DNA fingerprint and 16S ribosomal gene sequence analyses. FEMS Microbiol Lett. 2003;228:249–57. DOIPubMedGoogle Scholar
- Rudenko N, Golovchenko M, Hönig V, Mallátová N, Krbková L, Mikulásek P, et al. Detection of Borrelia burgdorferi sensu stricto ospC alleles associated with human lyme borreliosis worldwide in non-human-biting tick Ixodes affinis and rodent hosts in Southeastern United States. Appl Environ Microbiol. 2013;79:1444–53. DOIPubMedGoogle Scholar
- Rudenko N, Golovchenko M, Grubhoffer L, Oliver JH Jr. Borrelia carolinensis sp. nov., a novel species of the Borrelia burgdorferi sensu lato complex isolated from rodents and a tick from the south-eastern USA. Int J Syst Evol Microbiol. 2011;61:381–3. DOIPubMedGoogle Scholar
- Rudenko N, Golovchenko M, Lin T, Gao L, Grubhoffer L, Oliver JH Jr. Delineation of a new species of the Borrelia burgdorferi Sensu Lato Complex, Borrelia americana sp. nov. J Clin Microbiol. 2009b;47:3875–80. DOIPubMedGoogle Scholar
- Casjens SR, Mongodin EF, Qiu WG, Dunn JJ, Luft BJ, Fraser-Liggett CM, et al. Whole-genome sequences of two Borrelia afzelii and two Borrelia garinii Lyme disease agent isolates. J Bacteriol. 2011;193:6995–6. DOIPubMedGoogle Scholar
- Mongodin EF, Casjens SR, Bruno JF, Xu Y, Drabek EF, Riley DR, et al. Inter- and intra-specific pan-genomes of Borrelia burgdorferi sensu lato: genome stability and adaptive radiation. BMC Genomics. 2013;14:693. DOIPubMedGoogle Scholar
- Casjens SR, Di L, Akther S, Mongodin EF, Luft BJ, Schutzer SE, et al. Primordial origin and diversification of plasmids in Lyme disease agent bacteria. BMC Genomics. 2018;19:218. DOIPubMedGoogle Scholar
- Casjens S, Eggers C, Schwartz I. Comparative genomics of Borrelia. In: Samuels DS, Radolf JD, editors. Borrelia: molecular biology, host interaction and pathogenesis. Norwich (UK): Horizon Scientific Press; 2010. p. 26–52.
- Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–6. DOIPubMedGoogle Scholar
- Tourand Y, Deneke J, Moriarty TJ, Chaconas G. Characterization and in vitro reaction properties of 19 unique hairpin telomeres from the linear plasmids of the lyme disease spirochete. J Biol Chem. 2009;284:7264–72. DOIPubMedGoogle Scholar
- Kingry LC, Batra D, Replogle A, Rowe LA, Pritt BS, Petersen JM. Whole genome sequence and comparative genomics of the novel Lyme borreliosis causing pathogen, Borrelia mayonii. PLoS One. 2016;11:
e0168994 . DOIPubMedGoogle Scholar - Comstedt P, Jakobsson T, Bergström S. Global ecology and epidemiology of Borrelia garinii spirochetes. Infect Ecol Epidemiol. 2011;1:9545. DOIPubMedGoogle Scholar
- Takano A, Nakao M, Masuzawa T, Takada N, Yano Y, Ishiguro F, et al. Multilocus sequence typing implicates rodents as the main reservoir host of human-pathogenic Borrelia garinii in Japan. J Clin Microbiol. 2011;49:2035–9. DOIPubMedGoogle Scholar
- Gatzmann F, Metzler D, Krebs S, Blum H, Sing A, Takano A, et al. NGS population genetics analyses reveal divergent evolution of a Lyme Borreliosis agent in Europe and Asia. Ticks Tick Borne Dis. 2015;6:344–51. DOIPubMedGoogle Scholar
- Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:
vey016 . DOIPubMedGoogle Scholar - Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73. DOIPubMedGoogle Scholar
- Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography finds its roots. PLOS Comput Biol. 2009;5:
e1000520 . DOIPubMedGoogle Scholar - Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51:492–508. DOIPubMedGoogle Scholar
- Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–7. DOIPubMedGoogle Scholar
- Muzaffar SB, Smith RP Jr, Jones IL, Lavers J, Lacombe EH, Cahill BK, et al. Trans-Atlantic movement of the spirochete Borrelia garinii: the role of ticks and their seabird hosts. In: Paul E, editor. Emerging avian disease. Studies in Avian Biology, Volume 42. Berkeley (CA): University of California Press; 2012. p. 23–30.
Figure
Cite This ArticleOriginal Publication Date: December 17, 2022
1Current affiliation: National Institutes of Health, Bethesda, Maryland, USA.
Table of Contents – Volume 29, Number 1—January 2023
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Natalie Rudenko, Biology Centre, Czech Academy of Sciences, Institute of Parasitology, Branisovska 31, 37005, Ceske Budejovice, Czech Republic
Top