Volume 29, Number 9—September 2023
Dispatch
Reoccurring Escherichia coli O157:H7 Strain Linked to Leafy Greens–Associated Outbreaks, 2016–2019
Cite This Article
Citation for Media
Abstract
Genomic characterization of an Escherichia coli O157:H7 strain linked to leafy greens–associated outbreaks dates its emergence to late 2015. One clade has notable accessory genomic content and a previously described mutation putatively associated with increased arsenic tolerance. This strain is a reoccurring, emerging, or persistent strain causing illness over an extended period.
Escherichia coli O157:H7 is estimated to cause ≈63,000 domestically acquired foodborne illnesses and 20 deaths in the United States each year (1). E. coli O157:H7 infections are typically associated with abdominal cramps, bloody diarrhea, and vomiting; however, a rare but serious condition called hemolytic uremic syndrome can develop, resulting in anemia and acute renal failure (2). Healthy cattle serve as the main reservoir for E. coli O157:H7, and contaminated food, water, and environmental sources, as well as contact with animals, have been the source of outbreaks of E. coli O157:H7 infections (3,4). More recently, contaminated leafy greens have been recognized as a major source of E. coli O157:H7 illnesses and outbreaks. In foodborne illness attribution estimates for 2020 based on outbreak data, 58.1% of E. coli O157:H7 illnesses were attributed to vegetable row crops, a category that includes leafy greens (https://www.cdc.gov/foodsafety/ifsac/annual-reports.html). During 2009–2018, a total of 32 confirmed or suspected outbreaks of E. coli O157:H7 infections linked to contaminated leafy greens occurred in the United States and Canada (5).
A large E. coli outbreak in late 2019, hereafter referred to as outbreak A, caused 167 cases, hospitalized 85 persons from 27 states, and was associated with the consumption of romaine lettuce from Salinas Valley, California, USA (https://www.cdc.gov/ecoli/2019/o157h7-11-19/index.html). We characterized isolates from outbreak A and highly related isolates by using a variety of molecular methods.
A query of the PulseNet database revealed 356 isolates related to the outbreak strain that had <15 core-genome multilocus sequence typing (MLST; cgMLST) allele differences (Table 1; Appendix 1 Table 1) (6). Of those, 302 isolates corresponded to human cases associated with 6 outbreaks spanning 3 years; dates of isolation ranged from September 27, 2016, to January 3, 2020. An additional 54 isolates were either clinical isolates not associated with a recognized outbreak (n = 14) or from environmental (n = 20), food (n = 8), or animal (n = 12) samples. Seven-gene MLST and Manning clade typing revealed all isolates were sequence type (ST) 11 and belonged to Manning clade 2 (Appendix 2). In silico PCR of the Shiga toxin (stx) genes revealed that all but 2 isolates contained stx2a, whereas 2 remaining isolates had no detectable stx genes. We generated a closed-reference genome, 2019C-3201 (Strain: PNUSAE020169; BioSample: SAMN10432148), using PacBio Sequel technology (https://www.pacb.com) and assembled with Flye version 2.6 (7). The sequence data assembled into a single complete chromosomal contig and 3 plasmids (Table 2).
We selected a subset of 245 isolates for further genomic analysis to more evenly sample across outbreaks and to reduce computational demands. Isolates were characterized by core genome MLST implemented in BioNumerics 7.6 (6) and high-quality single-nucleotide polymorphism (SNP; hqSNP) methods using Lyve-SET version 1.1.4f (9), using the chromosomal sequence of 2019C-3201 as a reference and the Lyve-SET presets for E. coli. Overall, hqSNP was more discriminatory, differentiating isolates by a median of 10 pairwise hqSNPs (0–39 SNPs), whereas cgMLST differentiated isolates by a median of 2 allele differences (0–8 alleles) (Table 1). This finding was foreseeable because hqSNP does not depend on a predefined scheme; therefore, intergenic SNPs between loci, multiple SNP differences within a given locus, or SNPs in loci not included in the cgMLST schema can result (9).
Figure 1

Figure 1. Tip-dated maximum clade credibility tree of 245 isolates of reoccurring Escherichia coli O157:H7 strain REPEXH02 linked to leafy greens–associated outbreaks, 2016–2019, generated in BEAST2 (https://www.beast2.org). Tips...
Time-tree analysis using BEAST version 2.6.3 (10) revealed the divergence of this strain into 2 clades that last shared a common ancestor around late 2015 (median December 19, 2015; 95% highest posterior density interval December 7, 2014–July 10, 2016) (Figure 1). After outbreak D in 2016, sequences corresponding to a given outbreak belonged to 1 of 2 clades; outbreaks B2 and C were associated with clade 1, and outbreaks A, B1, and B3 were associated with clade 2. Of note, outbreak A was traced to romaine lettuce from Salinas Valley, whereas traceback and sampling in outbreak B2 linked some illnesses to romaine lettuce from Santa Maria, California (https://www.fda.gov/food/outbreaks-foodborne-illness/investigation-summary-factors-potentially-contributing-contamination-romaine-lettuce-implicated-fall; https://www.fda.gov/food/outbreaks-foodborne-illness/outbreak-investigation-e-coli-romaine-salinas-california-november-2019). Lettuce from Salinas was not considered a source of any illnesses in outbreak B2. Environmental sampling in Santa Maria in 2019 yielded isolates clustering closely with outbreak B2 in the time tree.
We analyzed the closed reference sequence of 2019C-3201 using Prokka version 1.8 to enable SNP annotation (11). We examined output from Lyve-SET to determine the SNPs differentiating the 2 clades in our phylogenetic analysis (Appendix 1 Table 2). This work confirms a previous study reporting a nonsense mutation in the arsR gene, an arsenical resistance operon repressor (12). All clade 1 isolates in this study possess a G→A mutation resulting in a premature stop codon. This mutation could decrease the activity of this repressor and lead to constitutive expression of this operon. Agricultural soils and water sources can contain increased arsenic levels because of natural processes, industrial sources, or agricultural uses of arsenic, such as application of arsenic-containing herbicides, pesticides, or animal drugs (13). This mutation could provide an ecologic advantage in environments containing high levels of arsenic. This finding underscores the potential need to routinely screen enteric bacterial strains for heavy metal resistance determinants, as well as to consider heavy metal levels in soil as part of traceback investigations.
Figure 2
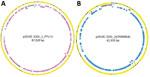
We further characterized isolates through assembly and annotation using Shovill-SPAdes version 1.0.9 and Prokka version 1.14.5 (11) and subsequent analysis in Roary version 3.11.2 (14) and scoary version 1.6.16 (15) to identify differences in the pangenome among isolates. We compared differentially distributed genes with the reference genome using BLASTn (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to identify feature location (chromosome/plasmid). Roary/scoary analysis revealed a subset of clade 1 isolates with additional genomic content. A total of 156 genomic features had >90 sensitivity and >90 specificity to this subset of clade 1. Of those, 87 (56%) are on plasmid p2019C-3201_1, and 69 (44%) are on p2019C-3201_2 (Figure 2; Appendix 1 Tables 3, 4). Prokka-annotated features associated with p2019C-3201_1 (Figure 2; Appendix 1 Table 3) were predominantly genes encoding hypothetical proteins with unknown functions and common plasmid-associated genes. Annotated features associated with p2019C-3201_2 (Figure 2; Appendix 1 Table 4) were predominantly associated with conjugation and span a large portion of that plasmid. Additional work is necessary to characterize the role of these plasmids in clade 1. When visualizing the distribution of these clade 1–specific features alongside the maximum-clade credibility tree (Figure 1; Appendix 1 Table 5), it appears those features were acquired after clade 1 and clade 2 diverged. Given the geographic distribution of isolates, these features might be a result of adaptation to a particular niche or environment.
In summary, a specific strain of E. coli O157:H7 associated with leafy greens has been the source of ongoing enteric illness since late 2016. This strain is estimated to have emerged in late 2015 and consists of 2 clades with different geographic distributions, 1 of which has notable genomic features. After this analysis, an additional outbreak associated with this strain was detected in late 2020 in which a reported 40 infections occurred in 19 states; 20 persons were hospitalized, and 4 developed hemolytic uremic syndrome (https://www.cdc.gov/ecoli/2020/o157h7-10-20b/index.html). After that outbreak, no further outbreaks have been detected, and only a single clinical isolate associated with this strain has been identified by PulseNet. The Centers for Disease Control and Prevention has classified this strain as a reoccurring, emerging, or persistent (REP) strain (https://www.cdc.gov/ncezid/dfwed/outbreak-response/rep-strains.html) with the designation REPEXH02. REP strains represent a new paradigm in enteric molecular surveillance, distinct from discrete outbreaks where numerous cases occur in a relatively short time frame. Detailed genomic characterization of additional REP strains, using the types of approaches outlined in this study, is necessary to elucidate factors contributing to their emergence and persistence in specific environments.
Dr. Chen is a bioinformatician in the Division of Foodborne, Waterborne, and Environmental Diseases, National Center for Emerging and Zoonotic Infectious Diseases, CDC. Her research interests are focused on understanding the evolution of bacterial foodborne pathogens of public health concern. Mr. Patel was an epidemiologist in Division of Foodborne, Waterborne, and Environmental Diseases, CDC, leading the response to multistate foodborne and zoonotic E. coli and Salmonella outbreaks within the Outbreak Response and Prevention Branch, as well as focusing on the response to REP strains of various pathogens. He now leads a team focused on building disease surveillance and outbreak response technology platforms in the private sector.
Acknowledgments
We thank state and local health departments for sequencing of E. coli O157:H7 associated with these outbreaks. The authors also thank Matthew Wise for helpful discussions and feedback.
This work was made possible by support from the Advanced Molecular Detection initiative at the Centers for Disease Control and Prevention and is covered by activities approved by the Centers for Disease Control and Prevention Internal Review Board (approval no. 7172).
References
- Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, et al. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis. 2011;17:7–15. DOIPubMedGoogle Scholar
- Bielaszewska M, Schmidt H, Liesegang A, Prager R, Rabsch W, Tschäpe H, et al. Cattle can be a reservoir of sorbitol-fermenting shiga toxin-producing Escherichia coli O157:H(-) strains and a source of human diseases. J Clin Microbiol. 2000;38:3470–3. DOIPubMedGoogle Scholar
- Heiman KE, Mody RK, Johnson SD, Griffin PM, Gould LH. Escherichia coli O157 Outbreaks in the United States, 2003-2012. Emerg Infect Dis. 2015;21:1293–301. DOIPubMedGoogle Scholar
- Marshall KE, Hexemer A, Seelman SL, Fatica MK, Blessington T, Hajmeer M, et al. Lessons learned from a decade of investigations of Shiga toxin–producing Escherichia coli outbreaks linked to leafy greens, United States and Canada. Emerg Infect Dis. 2020;26:2319–28. DOIPubMedGoogle Scholar
- Tolar B, Joseph LA, Schroeder MN, Stroika S, Ribot EM, Hise KB, et al. An overview of PulseNet USA databases. Foodborne Pathog Dis. 2019;16:457–62. DOIPubMedGoogle Scholar
- Lin Y, Yuan J, Kolmogorov M, Shen MW, Chaisson M, Pevzner PA. Assembly of long error-prone reads using de Bruijn graphs. Proc Natl Acad Sci U S A. 2016;113:E8396–405. DOIPubMedGoogle Scholar
- Redondo-Salvo S, Bartomeus-Peñalver R, Vielva L, Tagg KA, Webb HE, Fernández-López, et al. COPLA, a taxonomic classifier of plasmids. BMC Bioinfo. 2021;22:390. DOIGoogle Scholar
- Katz LS, Griswold T, Williams-Newkirk AJ, Wagner D, Petkau A, Sieffert C, et al. A comparative analysis of the Lyve-SET phylogenomics pipeline for genomic epidemiology of foodborne pathogens. Front Microbiol. 2017;8:375. DOIPubMedGoogle Scholar
- Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLOS Comput Biol. 2019;15:
e1006650 . DOIPubMedGoogle Scholar - Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. DOIPubMedGoogle Scholar
- Cherry JL. Recent genetic changes affecting enterohemorrhagic Escherichia coli causing recurrent outbreaks. Microbiol Spectr. 2022;10:
e0050122 . DOIPubMedGoogle Scholar - Punshon T, Jackson BP, Meharg AA, Warczack T, Scheckel K, Guerinot ML. Understanding arsenic dynamics in agronomic systems to predict and prevent uptake by crop plants. Sci Total Environ. 2017;581-582:209–20. DOIPubMedGoogle Scholar
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3. DOIPubMedGoogle Scholar
- Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016;17:238. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: August 16, 2023
1These first authors contributed equally to this article.
2Current affiliation: Tulane National Primate Research Center, Covington, Louisiana, USA.
Table of Contents – Volume 29, Number 9—September 2023
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Jessica C. Chen, Centers for Disease Control and Prevention, 1600 Clifton Rd NE, Mailstop H23-7, Atlanta, GA 30329-4027, USA
Top