Volume 3, Number 3—September 1997
Perspective
Resistance, Remission, and Qualitative Differences in HIV Chemotherapy
Cite This Article
Citation for Media
Abstract
To understand the role of qualitative differences in multidrug chemotherapy for human immunodeficiency virus (HIV) infection in virus remission and drug resistance, we designed a mathematical system that models HIV multidrug chemotherapy including uninfected CD4+ T cells, infected CD4+ T cells, and virus populations. The model, which includes the latent and progressive stages of the disease and introduces chemotherapy, is a system of differential equations describing the interaction of two distinct classes of HIV (drug-sensitive [wild type] and drug-resistant [mutant]) with lymphocytes in the peripheral blood; the external lymphoid system contributes to the viral load. The simulations indicate that to preclude resistance, antiviral drugs must be strong enough and act fast enough to drive the viral population below a threshold level. The threshold depends upon the capacity of the virus to mutate to strains resistant to the drugs. Above the threshold, mutant strains rapidly replace wild-type strains. Below the threshold, resistant strains do not become established, and remission occurs. An important distinction between resistance and remission is the reduction of viral production in the external lymphoid system. Also the virus population rapidly rebounds when treatment is stopped even after extended periods of remission.
Mathematical models provide a means to understand the human immunodeficiency virus (HIV)-infected immune system as a dynamic process. Models formulated as differential equations for the dynamic interactions of CD4+ lymphocytes and virus populations are useful in identifying essential characteristics of HIV pathogenesis and chemotherapy. Recent clinical studies have produced new insight into the dynamics of these virus populations during HIV infection (1-3). Turnover rates and lifespans of infected CD4+ T cells and virus have been identified by measuring their rates of change in patients undergoing strong antiviral monotherapy. The determination of these rates showed that large numbers of CD4+ T cells and virus are gained and lost each day throughout the course of HIV infection (4,5); these findings have profoundly influenced strategies for therapy (6). It is now recognized that chemotherapeutic agents must strongly suppress viral production before rapidly appearing viral mutants evolve to drug resistance. Recent clinical trials have accomplished this goal by using combined drug therapy. Ongoing trials with combinations of drugs have shown sharp declines, in some patients, of viral counts to nondetectable levels within several weeks of treatment; these levels were sustained for 1 year or more (5,7,8). At the same time, CD4+ T-cell counts have risen markedly before gradually leveling off (5,7,8). This apparent remission of HIV infection offers hope for the chronic control or even eradication of HIV (6). The issue of stopping treatment after such extended periods of remission, however, is yet to be resolved (8).
Our model of treatment distinguishes qualitatively two treatment outcomes indicated by clinical trials. The first is resistance. Examples of resistance for three-drug combined therapy are reported for completed clinical trials by Collier et al. (9). In these trials, there was on average an increase of CD4+ T-cell counts by approximately 30% (peaking at approximately 8 weeks and returning to baseline at approximately 40 weeks) and a decrease of plasma virus by approximately 70% (peaking at approximately 4 weeks and recovering to half baseline at approximately 40 weeks). The second treatment outcome is remission. Examples of remission are indicated in preliminary reports of ongoing clinical trials (5,7,8,10). In these trials, 1) plasma virus decreased sharply to nondetectable levels in 2 to 4 weeks, and these levels were sustained for periods of 1 year or more, and 2) CD4+ T-cell counts increased steadily by 100/mm3 or more before gradually leveling off to below normal levels (this below normal recovery is believed to be due to an impaired production of new CD4+ T cells from the thymus and other sources [11,12]; this assumption is incorporated into the model).
The model consists of differential equations for the variables T(t) (the CD4+ T-cell population uninfected by virus at time t), Ts(t) (the CD4+ T-cell population infected by drug-sensitive virus at time t), Tr(t) (the CD4+ T-cell population infected by drug-resistant virus at time t), Vs(t) (the drug-sensitive virus population at time t), and Vr(t) (the drug-resistant virus population at time t). All these virus populations reside in the circulating blood, in which the values of uninfected CD4+ T cells and virus can be clinically measured. The assumptions of the model and its equations are given in the Appendix
The model incorporates recent clinically determined dynamic information about the HIV-infected immune system. The essential elements are as follows. After an initial period of acute viremia in the first few weeks after seroconversion, CD4+ T-cell counts decline gradually from approximately 600 to 800/mm3 to 0/mm3 over approximately 10 years (11) (normal CD4+ T-cell counts are 800 to 1,200/mm3). The decline of CD4+ T cells is more rapid early in the infection (13). Infected CD4+ T cells constitute 4% or less of the CD4+ T-cell population (14). The half-life of an infected CD4+ T cell is approximately 2 days (1-3,6). After the initial viremia, plasma virus increases from below 50/mm3 to 1,000/mm3 or more during the variable course of infection with a sharp increase toward the end of the symptomatic phase (11). The lifespan of a virus outside the cell is about 7.2 hrs (1-3).
Figure 1

A typical untreated disease course based upon CD4+ T-cell counts and viral level is simulated in Figure 1a,b (the initial period of viremia is not included in the model). The simulation in Figure 1a,b is in close agreement with a typical disease course (11). The initial virus level is determined by the model's parameters, which do not change throughout the course of the disease. This assumption is consistent with recent clinical findings that disease prognosis is correlated to a set-point of virus level established in each patient soon after the initial viremia, and viral levels and replication rates remain relatively stable after the set-point (5,8,15,16). In the model, different set-points are obtained by varying key parameter values.
In the model, treatment is incorporated as the reduction of two separate rates. The reduction of these rates provides treatment control variables corresponding to the intensity and velocity of drug action. The variables are the rate at which virus infects uninfected CD4+ T cells and the rate of virus influx into the plasma from the external lymphoreticular system. Reduction of this second rate is the most important for treatment outcome, since it is believed that as much as 98% of the virus in the circulating blood is contributed by the external lymphoid compartment (5,8,17). In the simulations, the dynamics in the lymphoid compartment are modeled as a viral source term rather than mechanistically, since limited data are available for this compartment (18). Models of combined plasma-lymph compartment dynamics will appear in future work. When treatment begins, the model assumes that a proportion of drug-sensitive virus mutates to drug-resistant virus. This proportion is also a treatment control variable corresponding to the combination of drugs used or the presence of genetic diversity at different disease stages (19).
The model distinguishes primarily between resistance and remission in the assumption of a threshold condition for the virus population in the plasma (and thus for the virus population in the lymphoid compartment). The threshold condition is incorporated into the rate that controls the contribution of drug-resistant virus from the external lymphoid compartment to the plasma. When treatment drives the plasma virus level below the threshold, the drug-resistant virus population does not emerge, and the drug-sensitive virus population falls to near 0. This threshold cannot be reached simply by gradually lowering the drug-sensitive virus population. Two additional factors must be considered: 1) when the virus population is above threshold, the high mutational capacity and short lifespan of the virus results in rapid production of drug-resistant variants; and 2) as the virus population approaches the threshold, Darwinian competition gives competitive advantage to the resistant viral strain as the sensitive viral strain diminishes in fitness and in numbers.
To reach the threshold, the virus population must be brought down extremely fast before mutation and selection pressure allow resistant virus to propagate in the drug-altered environment. In the simulations, this rapid fall to the threshold can be achieved if treatment inhibits the rate of viral influx from the external lymphoid compartment sufficiently fast. The threshold value depends on the drugs used and the capacity of the virus to mutate against these drugs. In some patients, plasma levels were reduced by 99.9% or more, yet remission did not occur (1,2). In these cases, there may have been an extremely low threshold specific to the drugs used or a disproportionately lower suppression of virus in the lymphoid compartment than in the plasma.
Figure 2

Figure 5

Computer simulations of treatment are given in Figure 1c,d and Figures 2-5. The initial values at the start of treatment for the simulations are obtained from the simulation in Figure 1a,b. In all the simulations, the parameters are the same, except for the parameters controlling treatment and the resistance mutation.
In Figure 1c,d, resistance is simulated. The simulation corresponds to composite data given (9) for patients receiving the drug combination saquinavir, zidovudine, and zalcitabine (the first is a protease inhibitor and the other two are reverse transcriptase inhibitors). Despite an impressive increase in the CD4+ T-cell counts in Figure 1c (significantly higher than typically seen with zidovudine alone [20-23]), the reduction of viral influx from the external lymphoid compartment is not fast enough or strong enough to bring the virus below the threshold for remission. The viral level thus rebounds after a few weeks, and the T-cell population resumes a decline.
In Figure 2, resistance is simulated corresponding to data for patients receiving therapy with strong reverse transcriptase inhibitors (10). In these simulations, the reduction of the viral influx from the external lymphoid compartment is higher than in the simulation in Figure 1d, which means that the effect of the drug is stronger and the decrease of the virus level is faster. The resistance mutation parameter is also assumed to be higher than in Figure 1c,d, which means that resistance develops sooner. The exponential rates of increase of CD4+ T-cell counts in Figure 2a are inversely correlated to CD4+ T-cell starting values, as are the times to the appearance of resistance. The exponential rates of viral decay (Figure 2b), as indicated by the slopes of their logarithmic plots, are approximately parallel and thus not correlated to treatment starting values. This lack of correlation has been reported in clinical data (1,2).
In the simulations in Figure 2, the lack of correlation can be explained by the failure of the treatment to suppress rapidly enough the viral production caused by the external lymphoid system. As this production is suppressed at faster and faster rates, the viral exponential decay rates approach the actual loss rates of the plasma virus, which in this model are correlated to the CD4+ T-cell levels. The decay rates (Figure 2b) do not yield the actual half-life of free virus, which is shorter. The difference is due to the incomplete inhibition of the external lymphoid viral production. The exponential rate of viral decay in patients undergoing treatment is claimed to correspond to the decay of noninfectious virus produced by CD4+ T cells infected after treatment begins by infectious virus present before treatment begins (where it is assumed that after treatment begins, all newly produced virions are noninfectious) (3). It is claimed that the reciprocal of the viral decay rate is the average lifespan of infected CD4+ T cells (3). The model considered here has a different interpretation of the effects of treatment, since the production of virus from the external lymphoid compartment is not immediately blocked by treatment and thus influences the viral decay rate.
Figure 3
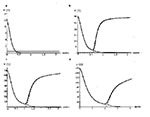
In Figure 3, simulations are given with the treatment parameter corresponding to suppression of virus influx from the lymphoid compartment higher than in Figure 1c,d and the mutation parameter lower than in Figure 2. In Figure 3a, remission is achieved, but in Figure 3b,c,d, it is not (the threshold value is indicated by the horizontal lines). The concurrence of strong suppression of the lymphoid virus compartment, lower resistance mutation parameter, and lower viral starting value allows the remission threshold to be reached (Figure 3a).
Figure 4

In Figure 4, treatment is simulated with an even higher value of the parameter corresponding to suppression of virus production from the external lymphoid compartment. Remission is achieved for the two lowest viral starting levels. As in Figure 2a, the exponential rates of increase of CD4+ T cells are inversely correlated to CD4+ T-cell starting values (an explanation in terms of the relative rates of changes in the differential equations for the CD4+ T-cell population is given in the Appendix). The exponential rates of viral decay (Figure 4b), however, are inversely correlated to increasing values of starting viral levels (in contrast with Figure 2b). When drug inhibition of virus in the lymph compartment is very high, the plasma viral clearance rate during treatment approaches the plasma viral clearance rate before treatment. In our models, it is assumed that the plasma viral clearance rate before treatment depends on CD4+ T-cell levels. The inverse correlation of plasma viral clearance rates during treatment to viral levels at the start of treatment is thus an indicator of higher viral clearance from the lymphoid compartment (an explanation in terms of the relative rates of change in the differential equation for the virus population is given in the Appendix).
In Figure 5a, treatment data for a patient receiving zidovudine, didanosine, and lamivudine are simulated (18; Figure 1d). This treatment induces a remission, even though the plasma virus does not fall below a nondetectable level. The plasma viral decay is approximately three times as fast as the lymph viral decay (18). This difference is incorporated into the treatment parameters for the simulation in Figure 5a. The two-phase plasma viral decay process (Figure 5a) matches the data (18) and is a strong indication that the rate of plasma viral decay is influenced by the slower rate of decay in the lymph system. In Figure 5b, the treatment simulation (Figure 5a) is stopped at 78 weeks, whereupon the drug-sensitive virus population rebounds sharply (8). This simulation is consistent with the report of a patient undergoing combined therapy who sustained nondetectable levels of virus for 78 weeks and upon voluntarily stopping treatment experienced high levels of virus in the blood within 1 week (3). In this simulation the virus would rapidly become reestablished even after much longer treatment. The resurgence of the virus population in the simulation is due to the capacity of the virus to grow very quickly from extremely low levels, which is due to the incomplete drug-induced inhibition of the external lymphoid viral source (18). This incomplete inhibition can be attributed to the presence of latently infected CD4+ T cells in the lymphoid compartment. Resumption of treatment in the simulation could again induce a remission. The resurgence of virus when treatment is stopped (Figure 5b) would hold also in all the simulations of remission because the 0 viral level is unstable; and if the virus is not suppressed by drugs, it rapidly grows from even very low levels. This instability of the 0 viral level results from the large viral influx from the lymph system, which is required to produce the characteristic dynamics of HIV throughout its entire progression.
Although computer models of HIV therapy are no substitute for clinical trials, they can bring into focus essential elements of the dynamic processes involved. The treatment simulations presented here identify the following qualitative dynamic elements involved in resistance and remission: 1) remission can occur if the viral production in the external lymphoid tissues is suppressed below a threshold level; 2) drug action must be strong enough and fast enough to drive the virus population to the threshold before resistant virus appears and propagates; 3) combination therapy or early treatment lowers the capacity of the virus to mutate to resistant strains and thus forestalls their emergence until the threshold is reached; and 4) stopping treatment even after an extended period of remission may result in a rapid rebound of the virus population.
The remission threshold is an abstract construct, and its quantitative value is relative to the capacity of the virus to mutate against a specific drug regimen. In the simulations, the threshold divides resistance and remission outcomes, and the dynamic developments in the first few days and weeks of drug administration are crucial in determining the outcome of therapy. Remission over extended time, however, may require continuing treatment to suppress low-level viral replication in the lymphoid tissues. The presence of even low-level viral production due to latently infected CD4+ T cells allows the possibility for the eventual evolution of drug-resistant viral strains.
Dr. Kirschner's research is in computational microbial pathogenesis in the Microbiology and Immunology Department, University of Michigan Medical School. Her work focuses on understanding the mechanisms for disease progression for such pathogens as HIV and Helicobacter pylori. She also studies the role of chemotherapy in disease dynamics as well as treatment strategies for these infections. Her recent work explores the role of the thymus in pediatric HIV infection.
Dr. Webb's research is in mathematical models of population dynamics; it includes theoretical investigations of differential equations that arise in models of biomathematics as well as computational investigations of specific models of biological populations, including models of the HIV-infected immune system, tumor growth, the spread of epidemics, and the blood production system.
References
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–6. DOIPubMedGoogle Scholar
- Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–22. DOIPubMedGoogle Scholar
- Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho D. HIV-1 Dynamics in vivo: clearance rate, infected cell lifespan, and viral generation time. Science. 1996;271:1582–6. DOIPubMedGoogle Scholar
- Piatak M, Saag MS, Yang LC, Clark SJ, Kappes JC, Luk KC, High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science. 1993;259:1749–54. DOIPubMedGoogle Scholar
- Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis and therapy. Science. 1995;267:483–9. DOIPubMedGoogle Scholar
- Stephenson J. New anti-HIV drugs and treatment strategies buoy AIDS researchers. JAMA. 1996;275:579–80. DOIPubMedGoogle Scholar
- Carpenter CJ, Fischl MA, Hammer SM, Hirsch MS, Jacobsen DM, Katzenstein DA, Antiretroviral therapy for HIV infection. JAMA. 1996;276:146–54. DOIPubMedGoogle Scholar
- Collier AC, Coombs RW, Schoenfeld DA, Bassett RL, Timpone J, Baruch A, Treatment of human immunodeficiency virus infection with saquinavir, zidovudine, and zalcitabine. N Engl J Med. 1996;334:1011–7. DOIPubMedGoogle Scholar
- Cohen J. Science. 1996;273:302.AIDS Conference. Chemokines share center stage with drug therapies. PubMedGoogle Scholar
- Pennisi E, Cohen J. Eradicating HIV from a patient: not just a dream? Science. 1996;272:1884. DOIPubMedGoogle Scholar
- Grody WW, Fligiel S, Naeim F. Thymus involution in the acquired immunodeficiency syndrome. Am J Clin Pathol. 1985;84:85–95.PubMedGoogle Scholar
- Philips AN, Sabin CA, Mocroft A, Janossy G. Antiviral therapy. Nature. 1995;375:195. DOIGoogle Scholar
- Embretson J, Zupancic M, Ribas JL, Burke RA, Racz P, Tenner-Racz K, Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–62. DOIPubMedGoogle Scholar
- Mellors JW, Rinaldo CR, Gupta P, White RM, Todd JA, Kingsely LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–70. DOIPubMedGoogle Scholar
- Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, HIV infection is active and progressive in lymphoid during the clinically latent stage of disease. Nature. 1996;362:355–8. DOIGoogle Scholar
- Lafeuillade A, Poggi C, Profizi N, Tamalet C, Costes O. Human immunodeficiency virus type I kinetics in lymph nodes compared with plasma. J Infect Dis. 1996;174:404–7.PubMedGoogle Scholar
- McLean A, Nowak M. Competition between AZT sensitive and AZT resistant strains of HIV. AIDS. 1992;6:71–9. DOIPubMedGoogle Scholar
- Fischl MA, Richmann DD, Hansen N, Collier AC, Carey JT, Para MF, The safety and efficiency of AZT in the treatment of subjects with mildly symptomatic HIV type 1. Ann Intern Med. 1990;112:727–37.PubMedGoogle Scholar
- Graham NMH, Zeger SL, Park LP, Vermund SH, Detels R, Rinaldo CR, The effects on survival of early treatment of HIV infection. N Engl J Med. 1992;326:1037–42.PubMedGoogle Scholar
- Hamilton JD, Hartigan PM, Simberkoff MS, Day PL, Diamond GR, Dickinson GM, A controlled trial of early vs late treatment with AZT in symptomatic HIV infection. N Engl J Med. 1992;326:437–43.PubMedGoogle Scholar
- Montaner JSG, Singer J, Schechter MT. Clinical correlates of in vitro HIV-1 resistance to zidovudine. Results of the Multicentre Canadian AZT trial. AIDS. 1993;7:189–95. DOIPubMedGoogle Scholar
- Kirschner DE, Webb GF. A model for treatment strategy in the chemotherapy of AIDS. Bull Math Biol. 1996;58:367–91. DOIGoogle Scholar
- Perelson AS, Kirschner DE, DeBoer R. The dynamics of HIV infection of CD4+ T cells. Math Biosci. 1993;114:81–125. DOIPubMedGoogle Scholar
- Kirschner DE, Webb GF. Effects of drug resistance on monotherapy treatment of HIV infection. Bull Math Biol 1997.
- Kirschner DE, Webb GF. A mathematical model of combined drug therapy of HIV infection. J Theor Med. 1997. In press.
Figures
Cite This ArticleTable of Contents – Volume 3, Number 3—September 1997
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Denise E. Kirschner, Department of Microbiology and Immunology, University of Michigan Medical School, 6730 Medical Science Building II, Ann Arbor, MI 48109-0620 USA;fax: 313-764-3562
Top