Volume 30, Number 6—June 2024
Dispatch
Encephalitozoon cuniculi Microsporidia in Cerebrospinal Fluid from Immunocompetent Patients, Czech Republic
Figure 1
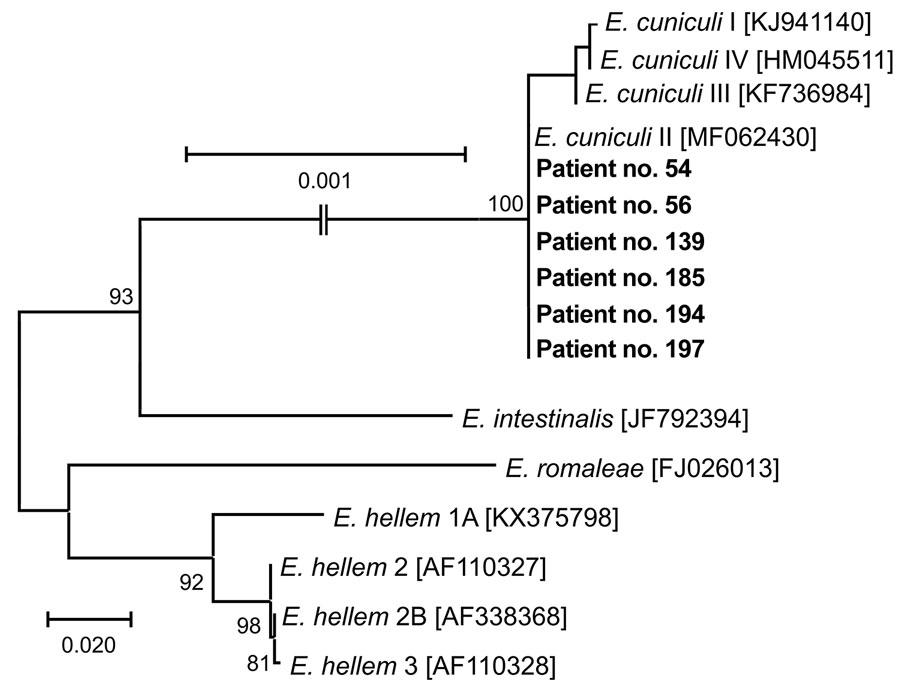
Figure 1. Phylogenetic analysis of Encephalitozoon cuniculi genotypes recovered from cerebrospinal fluid of immunocompetent patients, Czech Republic. Bold indicates sequences obtained in this study, identified by patient number. Sequences for comparisons were obtained from GenBank; accession numbers are in brackets. Tree was constructed by using the maximum-likelihood method. Partial sequences of 16S rRNA gene, the entire internal transcribed spacer region, and a partial sequence of 5.8S rRNA gene were inferred by using neighbor-joining analyses, and relationships were computed by using the Tamura 3-parameter method with gamma distribution and parametric bootstrap analysis of 1,000 replicates in MEGA X software (MEGA, https://www.megasoftware.net). Scale bar indicates nucleotide substitutions per site.