Volume 32, Number 5—May 2026
Dispatch
Retrospective Phylogenetic Analysis of Mayaro Virus, French Guiana, 1996–2024
Figure 2
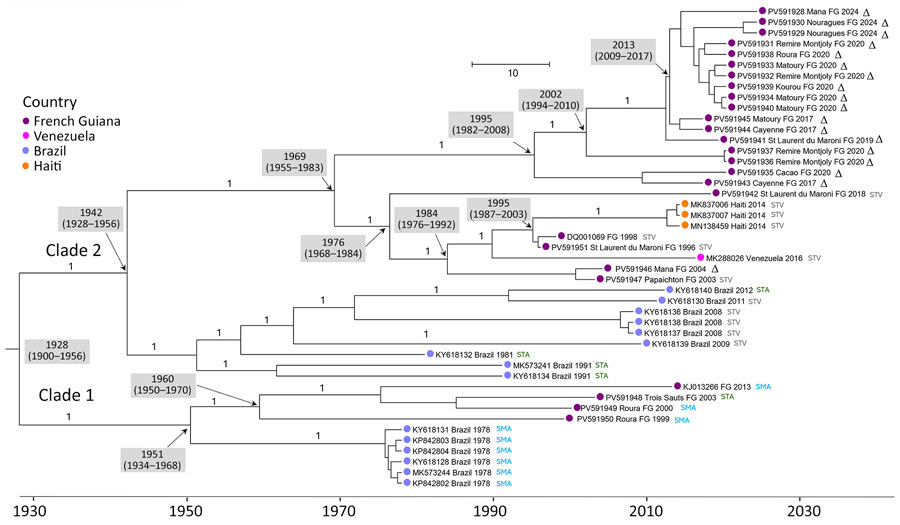
Figure 2. Bayesian phylogeny of 45 selected genotype D sublineage 2 coding sequences from a retrospective phylogenetic analysis of Mayaro virus, French Guiana, 1996–2024. Maximum clade credibility tree inferred by using the general time-reversible with gamma distribution and invariant site substitution model, under a strict clock and Bayesian skyline coalescent prior. The tree was generated by using TreeAnnotator version 1.10.4 (BEAST Developers, https://beast.community/treeannotator), and the resulting time-scaled phylogenies were visualized with FigTree version 1.4.3 (https://tree.bio.ed.ac.uk/software/figtree). Amino acid motifs at positions 1714–1716 are indicated by color at the terminal nodes of each sequence. Gray shaded boxes indicate dates of time to most recent common ancestor (95% highest posterior density). Bootstrap support values are indicated on the corresponding branches; a value of 1 corresponds to 100% bootstrap support. GenBank accession numbers are provided. Scale bar indicates nucleotide substitutions per site.
1These first authors contributed equally to this article.