Volume 8, Number 11—November 2002
Research
Molecular Analysis of Sarcoidosis Tissues for Mycobacterium Species DNA
Cite This Article
Citation for Media
Abstract
We performed polymerase chain reaction analysis, for Mycobacterium species 16S rRNA, rpoB, and IS6110 sequences, on 25 tissue specimens from patients with sarcoidosis and on 25 control tissue specimens consisting of mediastinal or cervical lymph nodes and lung biopsies. Mycobacterium species 16S rRNA sequences were amplified from 12 (48%) rpoB sequences from 6 (24%) of the sarcoidosis specimens. In total, 16S rRNA or rpoB sequences were amplified from 15 sarcoidosis specimens (60%) but were not detected in any of the control tissues (p=0.00002, Chi square). In three specimens, the sequences resembled Mycobacterium species other than M. tuberculosis. All specimens with sequences consistent with M. tuberculosis were negative for IS6110. We provide evidence that one of a variety of Mycobacterium species, especially organisms resembling M. tuberculosis, is found in most patients with sarcoidosis.
Sarcoidosis is a multisystem inflammatory disease that mainly affects lymph nodes and pulmonary tissues and is characterized by noncaseating granulomata in affected organs (1). Although the cause of sarcoidosis remains unknown, several microorganisms have been proposed as possible etiologic agents, including bacteria (Borrelia burgdorferi, Proprionibacterium acnes, and Mycobacterium species) and viruses (Human herpesvirus 8, Epstein-Barr virus, Cytomegalovirus, and Coxsackie B) (2). Metals (beryllium and zirconium), minerals (talc and clay), and organic substances (pine tree pollen) have also been proposed as etiologic agents (2). Efforts to identify an infectious agent for sarcoidosis using methods such as histologic staining and routine microbial culture have been unsuccessful.
Polymerase chain reaction (PCR) analysis for microbial DNA serves as an alternative method for identifying infectious agents. PCR was used to identify the etiologic agents of bacillary angiomatosis (Bartonella henselae) (3) and Whipple’s disease (Tropheryma whippelii) (4). Because of the substantial pathologic (5), immunologic (6), epidemiologic (7), and clinical similarities (8,9) between sarcoidosis and infections caused by Mycobacterium species (particularly tuberculosis), we analyzed tissue specimens from patients with sarcoidosis for evidence of mycobacterial genes. The results of previous studies have been inconclusive; some investigators were unable to demonstrate mycobacterial DNA in sarcoid lesions (10,11), whereas others have amplified mycobacterial DNA of different species (12,13). We examined sarcoidosis and control paraffin-embedded pulmonary, mediastinal, and cervical tissue specimens for Mycobacterium species 16S rRNA, rpoB, and IS6110 sequences.
Patients and Samples
For this study, we selected paraffin-embedded tissue specimens from patients who had had mediastinal or cervical lymph node resection from 1991 to 2001. Specimens from 44 patients with sarcoidosis and 57 controls were included. Patients were included for further study if they met the pathologic and clinical features described and if the specimens, after processing and DNA extraction, were positive for human β-actin with PCR analysis. We were unable to retrieve purified protein derivative status on a systematic basis. Based on these criteria, 25 control and 25 sarcoidosis specimens were further analyzed.
For inclusion in this study, the following criteria was used for patients with sarcoidosis: 1) clinical features had to be consistent with sarcoidosis (i.e., acute respiratory illness accompanied by erythema nodosum, hilar adenopathy and arthritis [Lofgren’s syndrome], or indolent progressive pulmonary decompensation associated with radiographic findings, such as hilar adenopathy, reticulonodular infiltrates, or pulmonary fibrosis); 2) histopathologic features had to be consistent with sarcoidosis (i.e., specimens from each patient had to have confluent noncaseating granulomas, well circumscribed within the surrounding tissue with a variable amount of peripheral lymphocytic infiltration [5]); 3) known microbial causes for granulomata had to be excluded (i.e., specimens were negative for microorganisms by hematoxylin and eosin (H&E), fungal, acid fast bacilli (AFB), and auramine-O stains and on routine bacterial, fungal, and AFB cultures). In each case, histopathologic specimens were independently reviewed by two pathologists.
Control lymph node specimens were selected from patients who had undergone mediastinoscopy or cervical node biopsy during the same period. In each case, a definitive diagnosis other than sarcoidosis was made. Control patients were selected from patients for whom the final diagnoses were fungal infection, lymphoma, and primary or metastatic lung malignancies (Table 1).
DNA Extraction
For each patient enrolled in the study, the original paraffin-embedded tissue block was retrieved from the archives, and eleven 10-µm sections were cut from each. One section was stained with H&E for microscopic examination, four sections were used for extraction of DNA, and the remaining six sections were stored for future analysis. The specimens were randomly processed for slide preparation, and the microtome blade was changed between each tissue block. For each section from patients with sarcoidosis and for the control specimens with the granulomata, granulomata were microdissected and extracted by using disposable surgical blades. For those control specimens without granulomata, all tissue from the four sections was used for DNA extraction. For all specimens, DNA was extracted with the Qiagen DNAeasy extraction kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions, except that 60 µL of proteinase K was used at a concentration of 20 mg/mL. Tissue dissection and DNA preparation were performed in a dedicated clean room, which was separate from the rooms used for PCR analysis and sequencing. The extracted DNA was stored at –20°C. Groups of tissue specimens from patients with sarcoidosis and controls were processed in parallel during all steps of the procedure, including extraction of the DNA, amplification and detection of mycobacterial DNA, and sequence analysis.
PCR Analysis for 16S rRNA, rpoB, and IS6110
Before PCR amplification, to ensure that the extracted DNA was of proper quality, we used PCR to verify that DNA sequences encoding human β-actin could be amplified. The primers used were 5´ATCATGTTTGAGACCTTCAAC3´ (forward primer) and 5´CAGGAAGGAAGGCTGGAAGAG3´ (reverse primer). The PCR conditions were 35 cycles of amplification carried out in a DNA Thermal Cycler 480 (Perkin-Elmer, Wellesley, MA); each cycle consisted of 1 min of denaturing at 94°C, 1 min of annealing at 54°C, and 1 min of extension at 72°C (14). As required, all 50 tissue specimens yielded human β-actin amplicons and were tested further for the presence of bacterial DNA.
For amplification of 16S rRNA sequences, a nested PCR analysis was performed. The primers FO16S, 5´GATAAGCCTGGGAAACTGGGTC3´ and RO16S, 5´TTCTCCACCTACCGTCAATCCG3´ were selected to amplify a 344-bp fragment of the 5´ region (nt 134–477) of mycobacterial 16S rRNA. Primers FI16S (5´CATGTCTTGTGGTGGAAAGCG3´) and RI16S (5´TACCGTCAATCCGAGAGAACCC3´) were selected as nested primers to amplify a 288-bp fragment (nt 181–468). The PCR conditions for both sets of primers were as follows: 5 min of denaturing at 94°C, followed by 35 cycles of amplification, consisting of 1 min of denaturing at 94°C, 1 min of annealing at 58°C, and 1 min of extension at 72°C. At the end of the 35 cycles, a final extension cycle of seven minutes at 72°C was performed.
For amplification of rpoB sequences, a nested PCR also was performed. The primers FOrpoB (5´GCAGACGCTGTTGGAAAACTTG3´) and ROrpoB, (5´TGTTCTGGTCCATGAATTGGCTC3´) were selected to amplify a 455-bp fragment of the β subunit (nt 1,940–2,394) of the M. tuberculosis RNA polymerase gene. The inner primers were designed as previously described and used in a nested fashion (nt 1,965–2,324) to amplify a 360-bp product (15). The PCR conditions were as described previously for 16S rRNA.
For amplification of IS6110 sequences, PCR analysis included the use of primers IS1 (5´CCTGCGAGCGTAGGCGTCGG3´) and IS2 (5´CTCGTCCAGCGCCGCTTCGG3´), designed to amplify a 123-bp fragment (nt 1,510–1,632) of the M. tuberculosis IS6110 element (16). The assay used the same conditions as described previously, with the exception that the PCR analysis included 30 rather than 25 cycles.
Negative and positive controls were run in parallel with each PCR assay. We used genomic DNA extracted from M. tuberculosis strain H37rv as positive controls, and DNA extracted from a paraffin-embedded tissue biopsy from an AIDS patient with ileocecal tuberculosis. We included the following as negative controls in each PCR reaction: DNA extracted from nonsarcoid paraffin-embedded tissue, PCR master mix inoculated with 5 µL of sterile water, and PCR master mix alone. The DNA extraction was performed in the same manner as described for the sarcoid and control specimens.
Determination of DNA Sequence of Amplified Products
The PCR products were purified with the Qiagen QIAquick PCR purification kit (Qiagen, Valencia, CA) and sequenced directly on both strands in the Vanderbilt Cancer Center Core Sequencing Laboratory. In cases in which the signal was ambiguous, PCR products were cloned into the plasmid vector system, pGEM T-Easy (Promega, Madison, WI), and the nucleotide sequences were then determined.
Alignments of the 16S rRNA, rpoB, and IS6110 sequences were performed with the NCBI BLAST program. Statistical evaluation of significance was determined by using Chi-square analysis or Fisher’s exact test, depending upon anticipated cell size. Sequences were aligned with ClustalW and subjected to phylogenetic analysis with HKY85 distance matrices with Paup 4.0b8 (Sinauer Associates, Sunderland, MA).
Patient and Specimen Characteristics
Of the 25 patients with sarcoidosis, 12% were African American and 88% Caucasian; 36% were men, and 64% were <50 years of age. No specimens were obtained from persons <18 years of age (Table 1). The control population was 20% African American and 80% Caucasian; 68% were men. Most (76%) of the control patients were >50 years of age; the age and sex of the control patients reflect the patient population undergoing mediastinoscopy to obtain a tissue diagnosis for probable malignancy. The control population consisted of patients with lung cancer (72%), chronic fungal infections (16%), or lymphoma (12%) (Table 1). Mediastinal lymph nodes were the source of specimens from 88% and 84% of the sarcoid and control patients, respectively. The remaining specimens from each group were obtained from either pulmonary or cervical lymph node biopsies. Granulomas were present in all of the sarcoidosis tissue specimens and in 7 of the 25 control specimens.
PCR Assay Sensitivities
The sensitivity of the PCR assay for each gene was determined by PCR analysis of serially diluted genomic DNA from M. tuberculosis strain H37rv, ranging from 5 ng to 0.05 fg per µL. One M. tuberculosis genome is estimated to have a mass of 5 fg (17). For PCR analyses of 16S rRNA, rpoB, and IS6110, we consistently achieved a sensitivity of 1–2 gene copies in each assay.
16S rRNA PCR of Tissue Specimens
Figure
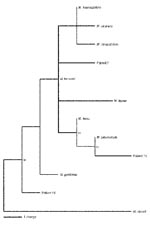
Figure. Analysis of 16S rRNA sequences from nine Mycobacterium species and from 12 patients with sarcoidosis. Phylograms based on nucleotide alignments were generated with HKY85 distances matrices using Paup 4.0b10 (Sinauer Associates,...
In the PCR for 16S rRNA sequences (Table 2), 12 (48%) of the 25 sarcoidosis specimens tested positive compared to none of 25 of the control specimens (p=0.0003, Chi square). Sequence analysis of the PCR products from the sarcoidosis specimens showed that 8 of the 12 had 100% positional identity with M. tuberculosis, and 1 possessed 99% positional identity with M. tuberculosis (patient 15). Sequencing of the 16S PCR product of patient 15 showed a C®T substitution at position 289 and an A®G substitution at position 355 (based on the M. tuberculosis 16S rRNA sequence, GenBank accession nos. Z83862.1, AJ131120.1, X52917.1, and X58890.1). Three other sequences were found (in patients 7, 19, and 24) that most closely resembled other Mycobacterium species. The amplicon sequence from patient 7 possessed an A®G substitution at position 299 and a C®A substitution at position 380, yielding 99% positional identity with M. kansasii. Notably the sequences of M. kansasii, M. avium, M. visibilis, and M. paratuberculosis are identical within this region; therefore, distinguishing between these species is not possible (18). The amplicon sequence from patient 19 contained a T®C substitution at position 434, yielding 99% positional identity with M. gordonae. The amplicon sequence from patient 24 contained 100% positional identity with M. gordonae and M. bohemicum (19). The phylogenetic relationships of the mycobacterial sequences are shown in the Figure and are deposited in GenBank (accession nos. AF468214, AF468215, and AF468216).
PCR of Tissue Specimens with Other Mycobacterial Primers
With the rpoB PCR, 6 (24%) of the 25 specimens from sarcoidosis patients yielded a product of 360 bp, which by sequence analysis in each case was most consistent with M. tuberculosis. Five sequences amplified from sarcoidosis specimens had 98%–100% positional identity with M. tuberculosis (patients 1, 3, 8, 14, and 24), whereas one had 95% positional identity (patient 16) (Table 2). The difference in the rate of finding M. tuberculosis rpoB sequences (24% in the sarcoidosis specimens and none in the control specimens) was also significant (p=0.02, Fisher’s exact test). In total, 15 (60%) of the sarcoidosis specimens had either Mycobacterium 16S rRNA or rpoB sequences compared with none of the control specimens (p=0.00002, Chi square). Mycobacterial 16S rRNA and rpoB fragments were amplified from 3 (12%) of the 25 sarcoidosis specimens (patients 1, 14, and 24). Patients 1 and 14 possessed sequences that had 100% positional identity with M. tuberculosis 16S rRNA and 98%–99% positional identity with M. tuberculosis rpoB DNA. The products amplified from patient 24 possessed 100% positional identity with M. gordonae 16S rRNA and 99% positional identity with M. tuberculosis rpoB DNA. In the region amplified with our 16S primers, a difference of 14 nt existed between the 16S rRNA of M. tuberculosis and M. gordonae. In the region amplified by using rpoB primers, a difference of 39 nt existed between rpoB of M. tuberculosis and M. gordonae.
None of the sarcoidosis or control patient tissue specimens yielded IS6110 amplicons (Table 2). In contrast, IS6110 products were consistently found in the positive controls, genomic DNA from M. tuberculosis H37rv, and DNA extracted from a paraffin-embedded tissue biopsy of an AIDS patient with ileocecal tuberculosis. Both positive controls were positive in the 16S rRNA, rpoB, and IS6110 PCR assays, and sequence analysis of the products indicated 100% homology with M. tuberculosis. The negative controls were consistently negative.
For this study, we chose patients whose cases were consistent with sarcoidosis or in whom an alternative diagnosis was made conclusively. We chose this stringent design so that no borderline tissues were examined. Many cases of disease eventually diagnosed as sarcoidosis have atypical findings. If our present observations are confirmed, such cases will be important for future analyses.
We found evidence of mycobacterial DNA in the granulomas of 24% of sarcoidosis specimens when assessing for rpoB, 48% in the same population when assessing for 16S rRNA; in total, 60% were positive for either. We acknowledge the limitations of studying archival tissue and the possibility of contamination; however, control tissues did not demonstrate positive results, making contamination less likely. Earlier studies have identified the presence of mycobacterial DNA in sarcoidosis tissue specimens with 30%–50% prevalence (12,13,20). Instead of a single organism being present, we provide evidence for a heterogenous population of Mycobacterium species in the sarcoidosis tissue specimens studied. Although we found evidence of organisms resembling M. tuberculosis, M. gordonae, and M. kansasii, other studies also have identified M. avium sequences (12,13).
We also provide DNA sequence evidence for novel mycobacteria in patient 15. Although most DNA sequences from the study patients most closely resemble M. tuberculosis, sequences resembling other mycobacterial species also were identified (Table 2, Figure). In several previous studies, non-tuberculosis mycobacteria also have been reported (13). One novel sequence is most closely related to 16S rDNA from M. tuberculosis, a known pulmonary pathogen, rather than to sequences from other mycobacterial species of lesser virulence. The consistent presence of two single polymorphisms in the same location in the novel sequence suggests a true polymorphism rather than an error introduced by Taq PCR. Moreover, the novel sequence was consistently absent from water, non-sarcoid paraffin-embedded tissue, and M. tuberculosis DNA controls. Synonymous substitutions in the M. tuberculosis genome are relatively rare, although genomic variations have been found in genes associated with antibiotic resistance (21). The DNA with the polymorphism suggests that a variant of M. tuberculosis, or a closely related novel mycobacterium, may be present in the sarcoidosis specimen.
The presence of M. tuberculosis DNA in 48% of sarcoidosis specimens is notable because clear clinical connections between sarcoidosis and tuberculosis have been made. On occasion, patients with documented tuberculosis develop sarcoidosis while on antituberculous treatment or vice versa (22–24). Mycobacterial DNA in sarcoidosis specimens may explain the clinical correlation between sarcoidosis and tuberculosis. That patients have developed sarcoidosis while on antituberculous therapy suggests that in those patients M. tuberculosis was not the etiologic agent of sarcoidosis. That 60% of the specimens we examined showed mycobacterial DNA agrees with certain previous studies (12,13), but other studies were negative for mycobacterial DNA (10,11). One possible explanation for these discordant results is that sarcoidosis represents one group of host responses to infectious agents of which mycobacteria represent the largest associated group. Alternatively, Mycobacterium species are present in many of the lesions but at extremely low levels, on either side of the threshold of detection. Such a hypothesis of small numbers of organisms provoking an intense inflammatory response, analogous to tuberculoid leprosy (25), could explain why organisms cannot be detected except by ultrasensitive methods. Yet another alternative explanation was our observation of degradation of the mycobacterial signal in the total DNA extract. We observed that mycobacterial DNA could be amplified from the positive specimens consistently over a 6–8 month period if the original DNA extract was maintained at –20oC. After this time period, fresh DNA extractions were necessary to demonstrate the presence of mycobacterial DNA. The original specimens, in which the mycobacterial DNA could no longer be amplified, remained positive for human β-actin by PCR analysis, although the band was weaker, suggesting either that the eukaryotic DNA degraded more slowly than prokaryotic DNA or that more signal was originally present. This degradation occurred despite minimizing freeze-thaws of extracted DNA and maintaining the DNA at –20°C. Our observation suggests that isolation of mycobacterial DNA from sarcoidosis specimens is best achieved by performing PCR analysis on fresh DNA extractions, which may help explain why other investigators had negative findings.
Based on these observations, we examined whether M. tuberculosis DNA was present in the sarcoid granuloma by testing for the presence of IS6110. PCR analysis for IS6110 is useful, since IS6110 is typically present in 1–25 copies in members of the M. tuberculosis complex. M. bovis BCG has only a single of copy of IS6110, whereas the higher copy numbers are typically found in M. tuberculosis isolates (26). We found no evidence of IS6110 DNA in our sarcoidosis or control tissue specimens.
Several possible explanations exist for the presence of mycobacterial 16S rRNA and rpoB, and the absence of IS6110 in the sarcoid specimens, although these three amplicons were consistently present in our positive controls. First, our assay for IS6110 may not have been sufficiently sensitive to detect the very low numbers of M. tuberculosis genomes in the sarcoidosis tissue specimens. In serial dilution studies, the assay was sensitive enough to detect one bacterial genome, comparable to results for the nested PCRs for 16S and rpoB. However, correlating the sensitivity of DNA extracted from bacterial culture to DNA extracted from formalin-fixed, paraffin-embedded tissue is not possible. Other laboratories that reported an assay sensitivity of 1–2 genome copies for IS6110 in sarcoidosis tissue extract were also unable to detect any IS6110 (11,27–29), which was consistent with our results. Studies assessing for IS6110 reflect a substantial portion of the literature that does not support the presence of mycobacterial DNA in the sarcoidosis tissue specimens (11,27,28,30).
A second possibility is that M. tuberculosis is present but the strains do not contain IS6110, since strains that possess one copy or no copies of IS6110 have been reported (31,32). In the United States, all of approximately 14,000 strains of M. tuberculosis tested have been shown to possess IS6110; some in low-copy number (33). Therefore, this scenario seems unlikely.
A third explanation is that while the agent we found associated with sarcoidosis has a close genetic relationship with M. tuberculosis, it is not M. tuberculosis. The genes for 16S and 23S are particularly suitable as targets for identifying microorganisms, since they are both well conserved and show variation indicative of their evolution and interrelationship with other organisms (34). This genetic variation is the basis for identifying the species of microorganisms in a particular genus, as this genetic variation is a constant property. Other members of the M. tuberculosis-complex (M. tuberculosis BCG, M. bovis, M. microti, and M. africanum) have 100% 16S and rpoB homology with M. tuberculosis but belong to different species; these strains are usually differentiated from M. tuberculosis by biochemical and clinical features. Although we could not attempt isolation of microorganisms from the formalin-fixed, paraffin-embedded specimens, future studies targeted to mycobacteria would be especially useful in confirming our observations and in characterizing any association with sarcoidosis.
We have found evidence for mycobacterial 16S rRNA and rpoB sequences in sarcoidosis tissue specimens but not in control tissue specimens. Upon sequence analysis, the products were most consistent with M. tuberculosis, but IS6110 could not be detected from these species. We also provide evidence of the presence of a heterogeneous mycobacterial population, including organisms highly related to M. tuberculosis, M. gordonae, and M kansasii. This heterogeneous population was found in individual sarcoidosis samples and, in one case, in the same sample (patient 24). These findings suggest that while M. tuberculosis and other Mycobacterium species may not be the sole microbial agents present in sarcoidosis tissues, they are commonly present and may play important roles. Further investigation into their presence and any putative etiologic agent is warranted.
Dr. Drake is supported in part by the Robert Wood Johnson Minority Medical Faculty Development Program, the Medical Research Service of the Department of Veterans Affairs, and by RO1 GM 63270 from the National Institutes of Health.
Dr. Drake is assistant professor of medicine at Vanderbilt University School of Medicine in the department of infectious diseases. Her interests include the investigation of possible infectious etiologic agents for chronic inflammatory states, such as sarcoidosis, by using molecular assays.
Acknowledgment
We thank David Relman for guidance throughout the project, Colorado State University (NIAID NO1 AI-75320) for supplying our laboratory with the Mycobacterium tuberculosis strain H37rv, and Edward McDonald for providing paraffin-embedded lymph node biopsy specimens.
References
- Crystal RG, Bitterman PB, Rennard SI, Hance AJ, Keogh BA. Interstitial lung diseases of unknown cause. Disorders characterized by chronic inflammation of the lower respiratory tract. N Engl J Med. 1984;310:154–66.PubMedGoogle Scholar
- American Thoracic Society (ATS). the European Respiratory Society (ERS) and the World Association of sarcoidosis and Other Granulomatous Disorders (WASOG). Statement on sarcoidosis. Am J Respir Crit Care Med. 1999;160:736–55.PubMedGoogle Scholar
- Relman DA, Loutit JS, Schmidt TM, Falkow S, Tompkins LS. The agent of bacillary angiomatosis. An approach to the identification of uncultured pathogens. N Engl J Med. 1990;323:1573–80.PubMedGoogle Scholar
- Relman DA, Schmidt TM, MacDermott RP, Falkow S. Identification of the uncultured bacillus of Whipple's disease. N Engl J Med. 1992;327:293–301.PubMedGoogle Scholar
- Scadding JG, Mitchell DN. Sarcoidosis. 2nd ed. London: Chapman and Hall; 1985. p 36–41.
- Foley N, Lambert C, McNicol M, Johnson N, Rook G. An inhibitor of tumour necrosis factor in the serum of patients with sarcoidosis, tuberculosis and Crohn’s disease. Clin Exp Immunol. 1990;80:395–9.PubMedGoogle Scholar
- Brett G. Epidemiological trends in tuberculosis and sarcoidosis in a district of London between 1958 and 1963. Tubercle. 1965;46:412–6. DOIGoogle Scholar
- Mitchell DN, Rees RJW. Some diseases of possible mycobacterial aetiology. In: Ratledge C, Stanford J, editors. The biology of mycobacteria. London: Academic Press; 1982. p. 525–36.
- Rook GA, Stanford JL. Slow bacterial infections or autoimmunity? Immunol Today. 1992;13:160–4. DOIPubMedGoogle Scholar
- Richter E, Greinert U, Kirsten D. Assessment of mycobacterial DNA in cells and tissues of mycobacterial and sarcoid lesions. Am J Respir Crit Care Med. 1996;153:375–80.PubMedGoogle Scholar
- Bocart D, Lecossier D, de Lassence A, Valeyre D, Battesti JP, Hance AJ. A search for mycobacterial DNA in granulomatous tissues from patients with sarcoidosis using the polymerase chain reaction. Am Rev Respir Dis. 1992;145:1142–8.PubMedGoogle Scholar
- Popper HH, Klemen H, Hoefler G, Winter E. Presence of mycobacterial DNA in sarcoidosis. Hum Pathol. 1997;28:796–800. DOIPubMedGoogle Scholar
- Li N, Bajoghi A, Kubba A, Bhawan J. Identification of mycobacterial DNA in cutaneous lesions of sarcoidosis. J Cutan Pathol. 1999;26:271–8. DOIPubMedGoogle Scholar
- Hall BL, Finn OJ. PCR-based analysis of the T-cell receptor V beta multigene family: experimental parameters affecting its validity. Biotechniques. 1992;13:248–57.PubMedGoogle Scholar
- Kim BJ, Lee SH, Lyu MA, Kim SJ, Bai GH, Chae GT, Identification of mycobacterial species by comparative sequence analysis of the RNA polymerase gene (rpoB). J Clin Microbiol. 1999;37:1714–20.PubMedGoogle Scholar
- Eisenach KD, Cave MD, Bates JH, Crawford JT. Polymerase chain reaction amplification of a repetitive DNA sequence specific for Mycobacterium tuberculosis. J Infect Dis. 1990;161:977–81.PubMedGoogle Scholar
- Del Portillo P, Munillo LA, Patarroyo ME. Amplification of a species-specific DNA fragment of Mycobacterium tuberculosis and its possible use in diagnosis. J Clin Microbiol. 1991;29:2163–8.PubMedGoogle Scholar
- Edwards U, Rogall T, Blocker H, Emde M, Bottger EC. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 1989;17:7843–53. DOIPubMedGoogle Scholar
- Rogall T, Wolters J, Flohr T, Bottger EC. Towards a phylogeny and definition of species at the molecular level within the genus Mycobacterium. Int J Syst Bacteriol. 1990;40:323–30.PubMedGoogle Scholar
- El-Zaatari FA, Naser SA, Markesich DC, Kalter DC, Engstand L, Graham DY. Identification of Mycobacterium avium complex in sarcoidosis. J Clin Microbiol. 1996;34:2240–5.PubMedGoogle Scholar
- Sreevatsan S, Pan X, Stockbauer KE, Connell ND, Kreiswirth BN, Whittam TS, Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissimenation. Proc Natl Acad Sci U S A. 1997;94:9869–74. DOIPubMedGoogle Scholar
- Wong CF, Yew WW, Wong PC, Lee J. A case of concomitant tuberculosis and sarcoidosis with mycobacterial DNA present in the sarcoid lesion. Chest. 1998;114:626–9. DOIPubMedGoogle Scholar
- Kent DC, Houk VN, Elliott RC, Sokolowski JW, Baker JH, Sorensen K. The definitive evaluation of sarcoidosis. Am Rev Respir Dis. 1970;101:721–7.PubMedGoogle Scholar
- Sieling RA, Modlin RL. Cytokine patterns at the site of mycobacterial infection. Immunobiology. 1994;191:378–87.PubMedGoogle Scholar
- van Soolingen D, Hermans PWM, De Haas PEW, Soll DR, Van Embden JDA. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis. J Clin Microbiol. 1991;29:2578–86.PubMedGoogle Scholar
- Wilsher ML, Menzies RE, Croxson MC. Mycobacterium tuberculosis DNA in tissues affected by sarcoidosis. Thorax. 1998;53:871–4.PubMedGoogle Scholar
- Vokurka M, Lecossier D, du Bois RM, Wallaert B, Kambouchner M, Tazi A, Absence of DNA from mycobacteria of the M. tuberculosis complex in sarcoidosis. Am J Respir Crit Care Med. 1997;156:1000–3.PubMedGoogle Scholar
- Klemen H, Husain AN, Cagle PT, Garrity ER, Popper HH. Mycobacterial DNA in recurrent sarcoidosis in the transplanted lung—a PCR-based study on four cases. Virchows Arch. 2000;436:365–9. DOIPubMedGoogle Scholar
- Chao SC, Yan JJ, Lee JY. Cutaneous sarcoidosis among Taiwanese. J Formos Med Assoc. 2000;99:317–23.PubMedGoogle Scholar
- Se Thoe SY, Tay L, Sng EH. Evaluation of amplicon- and IS6110-PCR for direct detection of Mycobacterium tuberculosis complex in Singapore. Trop Med Int Health. 1997;2:1095–101. DOIPubMedGoogle Scholar
- van Soolingen D, de Haas PE, Hermans PW, Groenen PM, van Embden JD. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis. J Clin Microbiol. 1993;31:1987–95.PubMedGoogle Scholar
- Bifani PJ, Shopsin B, Alcabes P, Mathema B, Kreiswirth BN, Liu Z, Molecular epidemiology and tuberculosis control. JAMA. 2000;284:305–7. DOIPubMedGoogle Scholar
- Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–40. DOIPubMedGoogle Scholar
Figure
Tables
Cite This ArticleTable of Contents – Volume 8, Number 11—November 2002
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Wonder Drake, Division of Infectious Diseases, Vanderbilt University Medical Center, A-3310 MCN, Nashville, TN 37232, USA; fax: 615-343-6160;
Top