Volume 11, Number 10—October 2005
Perspective
Antimicrobial Drug Resistance: "Prediction Is Very Difficult, Especially about the Future"1
Cite This Article
Citation for Media
Abstract
Evolution of bacteria towards resistance to antimicrobial drugs, including multidrug resistance, is unavoidable because it represents a particular aspect of the general evolution of bacteria that is unstoppable. Therefore, the only means of dealing with this situation is to delay the emergence and subsequent dissemination of resistant bacteria or resistance genes. Resistance to antimicrobial drugs in bacteria can result from mutations in housekeeping structural or regulatory genes. Alternatively, resistance can result from the horizontal acquisition of foreign genetic information. The 2 phenomena are not mutually exclusive and can be associated in the emergence and more efficient spread of resistance. This review discusses the predictable future of the relationship between antimicrobial drugs and bacteria.
Over the last 60 years, bacteria and, in particular, those pathogenic for humans have evolved toward antimicrobial drug resistance. This evolution has 2 key steps: emergence and dissemination of resistance.
Humans cannot affect emergence because it occurs by chance and represents a particular aspect of bacterial evolution. Emergence can result from mutations in housekeeping structural or regulatory genes or from acquiring foreign genetic information. However, much can be done to delay the subsequent spread of resistance. Dissemination can occur at the level of the bacteria (clonal spread), replicons (plasmid epidemics), or of the genes (transposons). These 3 levels of dissemination, which coexist in nature, are not only infectious but also exponential, since all are associated with DNA duplication. Clonal dissemination is associated with chromosome replication, plasmid conjugation with replicative transfer, and gene migration with replicative transposition (1). The spread of resistance has repeatedly been shown to be associated with antimicrobial drug use (2), which stresses the importance of the prudent use of these drugs; a notion reinforced by the observation that resistance is slowly reversible (3,4).
Therefore, attempting to predict the future of the relationship between antimicrobial drugs and bacteria is conceptually challenging and potentially useful. For the sake of convenience, the examples will be taken mainly from the work carried out in the author’s laboratory, although numerous other examples can be found in the literature.
The clinically relevant predictable resistance types are listed in the Table. Although they have not yet been reported, they may exist in nature; their apparent absence is, at least for some of them, rather surprising. For example, streptococci, including pneumococci and groups A, C, and G, can easily acquire in vitro conjugative plasmids from enterococci and stably maintain and phenotypically express them (5). Therefore, it is all the more surprising that genes commonly found on plasmids in the latter bacterial genus, such as bla for penicillinase production and aac6´-aph2´´ for resistance to nearly all commercially available aminoglycosides, have not yet emerged in streptococci. The situation is even more unusual for Listeria spp., which remain susceptible to most antimicrobial drugs even though they can acquire plasmids from both enterococci and staphylococci (6). However, the obligate intracellular existence of Chlamydia spp. likely protects them from contact with foreign DNA and accounts for their retained susceptibility to antimicrobial drugs.
Figure 1

One should distinguish "natural" antimicrobial drugs (e.g., kanamycin), which are produced by microorganisms from the environment, from semisynthetic (e.g., amikacin) and entirely synthetic compounds (e.g., quinolones), which are produced, at least in part, by humans. The microorganisms that produce natural antimicrobial drugs have to protect themselves from the products of their own secondary metabolism. To ensure their survival, these organisms have developed self-protection mechanisms similar to those found in resistant human pathogens (7); this observation led to the idea that the producers constitute the pool of origin of certain resistance genes (8). Therefore, the study of resistance in the strain used for the industrial production of an antimicrobial agent could allow a strong prediction about the mechanism that will be found later in bacteria pathogenic for humans. For example, the study of glycopeptide producers would have allowed the elucidation, long before it actually occurred, of the mechanism by which enterococci and, more recently, staphylococci could become resistant to these drugs (Figure 1).
As already noted, bacteria are resistant to antimicrobial drugs after horizontal DNA transfer or mutations. Thus, another prediction that can be made is that bacteria will transfer to susceptible species, resistance determinants already known in other bacterial genera, for example, the recent acquisition of glycopeptide resistance by Staphylococcus aureus from Enterococcus spp (9). However, this prediction is limited since it refers to mechanisms that have already been explained. In addition to being antimicrobial agent producers, the commensal bacteria of mammals, particularly those in the gut, also represent a pool of origin for resistance genes. When infections are treated with an antimicrobial agent, all bacteria in the host are affected, including the commensal flora, which could result in the selection of resistant commensals, particularly in children who are administered oral antimicrobial drugs too frequently. Large numbers of these resident bacteria are present in the digestive tract where they are often in transient, but intimate, contact with exogenous microorganisms that are in various developmental states, including competence. These conditions favor the transfer of genes by transformation and by conjugation. Including antimicrobial drugs in animal feed also leads to the selection of a pool of resistance genes that can be transferred to commensal bacteria in the human digestive tract and thus ultimately to human pathogens, even when selective pressure is absent (10).
In the case of mutations, predictions can be supported by 2 types of experimental approaches: in vivo with intact bacteria or in vitro by using DNA. Mutations resulting in resistance can be obtained in an accelerated fashion by using hypermutators, that is, bacteria deficient in the DNA repair system (11). Mutations are also accumulated by using continuous cultures, preferably in chemostats under suitable selective pressure. A similar enhanced rate of evolution can be obtained by (saturated) DNA mutagenesis, followed by transformation into an appropriate host. This technique, for example, was used successfully to study the extent of variations in penicillinase genes that generate extended-spectrum β-lactamase agents (12).
Modulation of Gene Expression
In addition to developing mutations in structural genes for drug targets, bacteria can become resistant after mutational events in motifs for gene expression, such as promoters (13), in regulatory modules, such as 2-component regulatory systems (14), or positioning upstream from a gene of a mobile (15,16) or stable (17) promoter. Enhanced expression of genetic information can also be caused by alterations in translation attenuation (18). The DNA regions involved in gene regulation are not always adjacent to the target gene. This factor makes finding regulatory mutations more complicated and makes detecting resistance by this mechanism generally impossible by genotypic techniques (19).
Dissemination by Transformation
Dissemination by transformation is more likely in spontaneously transformable bacterial species such as Streptococcus pneumoniae, Acinetobacter spp., and Neisseria spp. These bacteria can easily acquire, integrate, and express stretches of DNA. Since the latter can include portions of foreign chromosomes, this process renders chromosomal mutations infectious (20).
Combination of Mechanisms
Because of increased activity or the expanded spectrum of certain drug classes (e.g., β-lactam agents and fluoroquinolones) or of local therapy (e.g., extremely high concentrations in the gut after oral administration of glycopeptides that do not cross the digestive barrier) bacteria need to combine mechanisms that confer resistance to the same class of molecules. This process is necessary to achieve high-level resistance (21) or expand the substrate range provided by a single resistance mechanism (22). An example is provided by gram-negative bacteria and β-lactam agents. Extended-spectrum β-lactamase agents are point mutants of "old" penicillinases (23). Generally, the biologic price to pay for extending the substrate range of this enzyme is hypersusceptibility to β-lactamase inhibitors. However, the presence in certain enterobacteria of the gene for a penicillinase on a small multicopy plasmid, which results in production of large amounts of the enzyme and confers resistance to β-lactamase inhibitors by trapping (24). The net result of this combinatorial approach is the production of gram-negative bacteria that are resistant to all β-lactam agents, except carbapenems and cephamycins, which are not substrates for the enzymes.
Two Mechanisms Involved in Resistance Are Increasingly Frequent
Impermeability
No antimicrobial agent is active against all bacteria. In fact, the intrinsic (natural) resistance of bacteria, which is better designated as insensitivity, defines the spectrum of activity of a drug, usually because the antimicrobial drug does not penetrate the bacteria. However, microorganisms can become resistant to nearly all drug classes, including those that act at the surface of the bacteria (e.g., β-lactam agents, bacitracin), by impermeability. This resistance can be secondary to 2 distinct pathways: passive, which involves alterations of outer membrane proteins, the porins, which decrease the rate of entry of antimicrobial drugs into the bacteria by diminution of the pore size (25), and active, which involves overexpression of an indigenous efflux pump that exports the antimicrobial drug outside the cell after a regulatory mutation (26).
Trapping
Figure 2
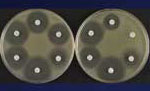
The mechanism of trapping, already mentioned in the case of resistance to β-lactam agents by a combination of β-lactamases, allows titration of the drugs, an alternative to impermeability, for lowering the intracellular concentrations of the antimicrobial drugs. This mechanism also works against aminoglycosides in bacteria that overproduce an enzyme that has affinity for a drug they cannot inactivate since it lacks the modification site (Figure 2) (27,28). This mechanism has also been proposed to account for low-level resistance to glycopeptides in staphylococci by overproducing target sites in the outer layers of the peptidoglycan; thus, the antimicrobial drug does not reach the important target sites where the wall is assembled on the outer surface of the cytoplasmic membrane (29).
Genes from gram-positive cocci can be transferred by conjugation (of plasmids or transposons) not only among these microorganisms but also to gram-negative bacteria (30). The reverse is not true because of limitations in heterologous gene expression. Consequently, one can confidently predict further dissemination of the resistance gene pool of gram-positive to gram-negative bacteria.
We have been aware for a long period that "everything that exists in the universe is the result of chance and necessity" (Democritus, 460–370 BC), which holds true for antimicrobial drug resistance. Most unfortunately, and for various reasons, it is extremely difficult to think like a bacterium. In other words, predicting the emergence of resistance to a drug class by a precise molecular mechanism is nearly impossible (e.g., glycopeptide resistance in enterococci or plasmid-mediated resistance to fluoroquinolones). We also cannot anticipate, among all the conceivable mechanisms of resistance (31), which will emerge first under natural conditions. However, based on the understanding during recent decades of the physiology (genetics and biochemistry) of bacterial resistance to antimicrobial drugs, impressive progress has been made in the techniques for in vitro detection and for elucidation of resistance. This progress should, in turn, be helpful in delaying the second step of resistance: dissemination.
Dr Courvalin is a professor at the Institut Pasteur, where he directs the French National Reference Center for Antibiotics and is the head of the Antibacterial Agents Unit. He specializes in the genetics and biochemistry of antimicrobial drug resistance.
Acknowledgments
I thank N. Bohr for providing the title, V. Loncle-Provot for inspiring me to write this review, and M.H. Saier for providing laboratory facilities.
This article is dedicated to the memory of my colleague and friend Maurice Hofnung.
References
- Courvalin P, Trieu-Cuot P. Minimizing potential resistance: the molecular view. Clin Infect Dis. 2001;33:S138–46. DOIPubMedGoogle Scholar
- Seppala H, Klaukka T, Lehtonen R, Nenonen E, Huovinen P. Outpatient use of erythromycin: link to increased erythromycin resistance in group A streptococci. Clin Infect Dis. 1995;21:1378–85. DOIPubMedGoogle Scholar
- Andersson DI. Persistence of antibiotic resistant bacteria. Curr Opin Microbiol. 2003;6:452–6. DOIPubMedGoogle Scholar
- Chiew YF, Yeo SF, Hall LM, Livermore DM. Can susceptibility to an antimicrobial be restored by halting its use? The case of streptomycin versus Enterobacteriaceae. J Antimicrob Chemother. 1998;41:247–51. DOIPubMedGoogle Scholar
- Macrina FL, Archer GL. Conjugation and broad host range plasmids in streptococci and staphylococci. In: Clewell DB, editor. Bacterial conjugation. New York, London: Plenun Press; 1993. p. 313–29.
- Charpentier E, Courvalin P. Antibiotic resistance in Listeria spp. Antimicrob Agents Chemother. 1999;43:2103–8.PubMedGoogle Scholar
- Mazodier P, Davies J. Gene transfer between distantly related bacteria. Annu Rev Genet. 1991;25:147–74. DOIPubMedGoogle Scholar
- Walker MS, Walker JB. Streptomycin biosynthesis and metabolism. J Biol Chem. 1970;245:6683–9.PubMedGoogle Scholar
- Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science. 2003;302:1569–71. DOIPubMedGoogle Scholar
- Moubareck C, Bourgeois N, Courvalin P, Doucet-Populaire F. Multiple antibiotic resistance gene transfer from animal to human enterococci in the digestive tract of gnotobiotic mice. Antimicrob Agents Chemother. 2003;47:2993–6. DOIPubMedGoogle Scholar
- Taddei F, Matic I, Godelle B, Radman M. To be a mutator, or how pathogenic and commensal bacteria can evolve rapidly. Trends Microbiol. 1997;5:427–8. DOIPubMedGoogle Scholar
- Petrosino J, Cantu C III, Palzkill T. β-lactamases: protein evolution in real time. Trends Microbiol. 1998;6:323–7. DOIPubMedGoogle Scholar
- Chen ST, Clowes RC. Variations between the nucleotide sequences of Tn1, Tn2, and Tn3 and expression of β-lactamase in Pseudomonas aeruginosa and Escherichia coli. J Bacteriol. 1987;169:913–6.PubMedGoogle Scholar
- Baptista M, Depardieu F, Reynolds P, Courvalin P, Arthur M. Mutations leading to increased levels of resistance to glycopeptide antibiotics in VanB-type enterococci. Mol Microbiol. 1997;25:93–105. DOIPubMedGoogle Scholar
- Goussard S, Sougakoff W, Mabilat C, Bauernfeind A, Courvalin P. An IS1-like element is responsible for high-level synthesis of extended-spectrum β-lactamase TEM-6 in Enterobacteriaceae. J Gen Microbiol. 1991;137:2681–7.PubMedGoogle Scholar
- Rudant E, Courvalin P, Lambert T. Characterization of IS18, an element capable of activating the silent aac(6´)-Ij gene of Acinetobacter sp. 13 strain BM2716 by transposition. Antimicrob Agents Chemother. 1998;42:2759–61.PubMedGoogle Scholar
- Magnet S, Courvalin P, Lambert T. Activation of the cryptic aac(6´)-Iy aminoglycoside resistance gene of Salmonella by a chromosomal deletion generating a transcriptional fusion. J Bacteriol. 1999;181:6650–5.PubMedGoogle Scholar
- Leclercq R, Courvalin P. Resistance to macrolides and related antibiotics in Streptococcus pneumoniae. Antimicrob Agents Chemother. 2002;46:2727–34. DOIPubMedGoogle Scholar
- Courvalin P. Genotypic approach to the study of bacterial resistance to antibiotics. Antimicrob Agents Chemother. 1991;35:1019–23.PubMedGoogle Scholar
- Ferrandiz MJ, Fenoll A, Liñares J, de la Campa AG. Horizontal transfer of parC and gyrA in fluoroquinolone-resistant clinical isolates of Streptococcus pneumoniae. Antimicrob Agents Chemother. 2000;44:840–7. DOIPubMedGoogle Scholar
- Arthur M, Courvalin P. Contribution of two different mechanisms to erythromycin resistance in Escherichia coli. Antimicrob Agents Chemother. 1986;30:694–700.PubMedGoogle Scholar
- Ferretti JJ, Gilmore KS, Courvalin P. Nucleotide sequence analysis of the bifunctional 6´-aminoglycoside acetyltransferase, 2´´-aminoglycoside phosphotransferase determinant from Streptococcus faecalis: identification and cloning of gene regions specifying the two activities. J Bacteriol. 1986;167:631–8.PubMedGoogle Scholar
- Sougakoff W, Goussard S, Gerbaud G, Courvalin P. Plasmid-mediated resistance to third-generation cephalosporins caused by point-mutations in TEM-type penicillinase genes. Rev Infect Dis. 1988;10:879–84. DOIPubMedGoogle Scholar
- Mabilat C, Courvalin P. Development of "oligotyping" for characterization and molecular epidemiology of TEM β-lactamases in members of the family Enterobacteriaceae. Antimicrob Agents Chemother. 1990;34:2210–6.PubMedGoogle Scholar
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. DOIPubMedGoogle Scholar
- Li XZ, Nikaido H. Efflux-mediated drug resistance in bacteria. Drugs. 2004;64:159–204. DOIPubMedGoogle Scholar
- Ménard R, Molinas C, Arthur M, Duval J, Courvalin P, Leclercq R. Overproduction of 3´-aminoglycoside phosphotransferase type I confers resistance to tobramycin in Escherichia coli. Antimicrob Agents Chemother. 1993;37:78–83.PubMedGoogle Scholar
- Magnet S, Smith TA, Zheng R, Nordmann P, Blanchard JS. Aminoglycoside resistance resulting from tight drug binding to an altered aminoglycoside acetyltransferase. Antimicrob Agents Chemother. 2003;47:1577–83. DOIPubMedGoogle Scholar
- Srinivasan A, Dick JD, Perl TM. Vancomycin resistance in staphylococci. Clin Microbiol Rev. 2002;15:430–8. DOIPubMedGoogle Scholar
- Courvalin P. Transfer of antibiotic resistance genes between gram-positive and gram-negative bacteria. Antimicrob Agents Chemother. 1994;38:1447–51.PubMedGoogle Scholar
- Quintiliani R Jr, Sahm D, Courvalin P. Mechanisms of resistance to antimicrobial agents. In Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH, editors. Manual of clinical microbiology. 7th ed. Washington: American Society for Microbiology; 1998. p. 1505–25.
- Pootoolal J, Thomas MG, Marshall CG, Neu JM, Hubbard BK, Walsh CT, Assembling the glycopeptide antibiotic scaffold: the biosynthesis of A47934 from Streptomyces toyocaensis NRRL15009. Proc Natl Acad Sci U S A. 2002;99:8962–7.PubMedGoogle Scholar
- Arthur M, Molinas C, Depardieu F, Courvalin P. Characterization of Tn1546, a Tn3-related transposon conferring glycopeptide resistance by synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J Bacteriol. 1993;175:117–27.PubMedGoogle Scholar
Figures
Table
Cite This Article1Niels Bohr.
Table of Contents – Volume 11, Number 10—October 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Patrice Courvalin, Unité des Agents Antibactériens, Institut Pasteur, 75015 Paris, France; fax: 00-1-45-68-83-19
Top