Volume 17, Number 3—March 2011
Dispatch
Phylogeny of European Bat Lyssavirus 1 in Eptesicus isabellinus Bats, Spain
Cite This Article
Citation for Media
Abstract
To better understand the epidemiology of European bat lyssavirus 1 (EBLV-1) in Europe, we phylogenetically characterized Lyssavirus from Eptesicus isabellinus bats in Spain. An independent cluster of EBLV-1 possibly resulted from geographic isolation and association with a different reservoir from other European strains. EBLV-1 phylogeny is complex and probably associated with host evolutionary history.
The genus Lyssavirus comprises 3 species that can infect bats in Europe: European bat lyssavirus 1 (EBLV-1), European bat lyssavirus 2, and West-Caucasian bat virus (1,2). Most lyssavirus-infected bats have been found in north-central Europe (Germany, the Netherlands, Denmark, Poland, and France); of these, >95% were serotine bats (Eptesicus serotinus) infected by EBLV-1 (3–5). EBLV-1 in other bat species has rarely been described (3,6). EBLV-1–infected bats become increasingly scarce from north to south in Europe, and no cases in northern Spain or Italy have been reported. The same trend has been consistently found within Germany (3) except for an artifact that arose from varied surveillance intensity among different countries. However, several infected serotine bats in southern Spain have been reported (7). These bats have been assigned to the species E. isabellinus, which has closely related populations on the African side of the Gibraltar Strait (8). This species is strongly divergent from E. serotinus bats (>16% of cytochrome b gene) in the northern Iberian Peninsula (9). In Spain, the distribution of EBLV-1 cases in bats apparently coincides with the distribution of E. isabellinus bats; 10 cases of human exposure after contact with infected bats have been reported; each was associated with E. isabellinus bats.
Two subtypes have been proposed for EBLV-1: EBLV-1a, which extends from the Netherlands to Russia in a west–east axis, and EBLV-1b, which includes strains that extend south through France and the Netherlands and the only 2 published strains from Iberia (1). We phylogenetically characterized EBLV-1 strains associated with E. isabellinus bats, a reservoir in the Iberian Peninsula that differs from E. serotinus bats.
We sequenced 12 bat brains positive for Lyssavirus antigen detected by immunofluorescence and reverse transcription–PCR (RT-PCR) as described (10). All viruses were identified as EBLV-1. For phylogenetic analyses, the 400-bp 5′ variable extreme of the nucleoprotein gene of these EBLV-1 strains was amplified by specific EBLV-1 nested RT-PCR and sequenced by using the following primers: SEQVAR1F 5′-1ACGCTTAACAACCAGATCAAAG22-3′, SEQVAR2F 5′-51AAAAATGTAACACYYCTACA70-3′, EBLVSEQVAR1R 5′-596CAGTCTCAAAGATCTGTTCCAT575-3′, and EBLVSEQVAR2R 5′-552TAGTTCCCAGTATTCTGTCC533-3′.
Figure 1

Figure 1. European bat lyssavirus 1 (EBLV-1) phylogenetic reconstruction based on the first 400 bp of the nucleoprotein gene. The tree was obtained by Bayesian inference run for 107 generations; trees were sampled...
All rabies-positive serotine bats came from southern Spain (Huelva, Seville, Murcia, and Badajoz) and were molecularly identified as E. isabellinus (8). An alignment was performed by using ClustalX (www.clustal.org) to combine the obtained sequences and other available EBLV-1 sequences from GenBank, including a Duvenhage virus used as the outgroup (online Appendix Table, www.cdc.gov/EID/content/17/3/520-appT.htm). Before conducting further analyses, we used jModelTest (http://darwin.uvigo.es/software/jmodeltest.html) to select the best fitting substitution model for our sequences according to the corrected Akaike information criterion. Maximum-likelihood phylogenies were reconstructed by using PHYML (http://atgc.lirmm.fr/phyml) software and by using a generalized time-reversible model and the γ parameter estimated in the analyses. Maximum-parsimony analyses were conducted by using PAUP* 4.0b10 (http://paup.csit.fsu.edu/) weighting transversions 15× according to the transitions/transversion ratio estimated in the jModelTest analyses. Confidence in the topologies for the maximum-likelihood and the maximum-parsimony analyses was established with 1,000 bootstrap replicates. A Bayesian phylogenetic inference was obtained by using MrBayes version 3.1 (http://mrbayes.csit/fsu.edu/) with random starting trees without constraints. Two simultaneous runs of 107 generations were conducted, each with 4 Markov chains, and the trees were sampled every 100 generations. Net p-distances between groups were calculated by using MEGA4 (www.megasoftware.net) (Figure 1).
The genetic structure and relationships between haplotypes were examined within the main lineages through a parsimony-based network built with a median-joining algorithm implemented in the Network 4.5.1 program (11). To evaluate and compare genetic variability and polymorphism among lineages, we estimated the number of haplotypes, mutations, and segregating sites as well as haplotype diversity and nucleotide diversity by using DNAsp version 4.5 (12) for the major clades (Table). Finally, to investigate population dynamics across lineages, the Fu Fs and Tajima D statistics were calculated (Table). These 2 statistics are considered to be the most powerful tests for detecting expansion events (13).
Figure 2
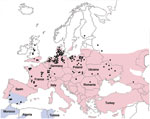
Figure 2. Geographic distribution of Eptesicus serotinus bats (red), E. isabellinus bats (blue), and cases of rabies in bats (green dots), Europe, 1990–2009. Obtained from Rabies Bulletin Europe (www.who-rabies-bulletin.org/).
All phylogenetic analyses, regardless of the reconstruction criterion used, formed a monophyletic cluster of the EBLV-1 strains from Spain (only the Bayesian inference reconstruction is shown). The Bayesian inference, maximum-likelihood, and maximum-parsimony analyses identified the cluster from Spain and EBLV-1a and EBLV-1b as being monophyletic (Figure 1), although only maximum-likelihood and maximum-parsimony analyses suggested a closer relationship between EBLV-1a and the cluster from Spain. The genetic differentiation of the EBLV-1 strains from the Iberian Peninsula matches their association with another bat species (Figure 2), which suggests that the host bat’s evolutionary history plays a major role in EBLV-1 molecular epidemiology, as has been proposed for rabies virus in bats in North America (14).
The low genetic diversity and the Fu Fs and Tajima D statistics (Table) all suggest rapid population expansion of EBLV-1a, which is consistent with the star-like structure of the network for this lineage (Figure 1). Conversely, haplotype and nucleotide diversity descriptors (Table) have the highest values for EBLV-1b and a complex network structure with differentiated subnetworks. All these elements indicate that this lineage has a complex evolutionary history. The lineage from Spain also has low diversity and a star-shaped network, but neutral evolution cannot be rejected on the basis of the Fs and D statistics. Net distances are similar within and between lineages, except for EBLV-1a, which is slightly more differentiated (Figure 1). Consequently, the suggested EBLV-1 expansion from Spain into Europe (15) is not supported by our results, which record the highest variability and most complex phylogenetic structure for France and the Netherlands (Figure 1). This complex structure suggests either a longer evolutionary history in these areas or a recent contact of distinct bat lineages in this zone.
The results of this study show that the strains from Spain do not belong to subtype 1b because of their association with a different reservoir (E. isabellinus bats). Moreover, what is currently considered to be EBLV-1b seems to include at least 4 lineages that are more genetically diverse and have a complex history. EBLV-1a, however, has low genetic diversity despite its extensive geographic distribution, suggesting a relatively recent and successful expansion of this lineage. These results call into question the current classification of EBLV-1 into 2 single subtypes. To provide a better understanding of EBLV-1 molecular epidemiology in Europe, additional studies that consider different genes should be conducted and the current classification should be revised accordingly.
Dr Vázquez-Morón is a PhD candidate at the Instituto de Salud Carlos III and Complutense University of Madrid. Her main research interests are the epidemiology and public health implications of rabies and emerging viruses in bats.
Acknowledgments
We thank the Genomics Unit of the Instituto de Salud Carlos III for analyses of the genomic sequences and Enrique Royuela Casamayor for his involvement in the daily work.
This project was financially supported by an agreement between the Public Health Department of the Spanish Ministry of Health and the Instituto de Salud Carlos III for the development of “Rabies Surveillance in Spain” and by projects SAF 2006-12784-C02-01 and SAF 2006-12784-C02-02 of the General Research Programme of the Spanish Ministry of Science and Education.
References
- Amengual B, Whitby JE, King A, Cobo JS, Bourhy H. Evolution of European bat lyssaviruses. J Gen Virol. 1997;78:2319–28.PubMedGoogle Scholar
- Botvinkin AD, Poleschuk EM, Kuzmin IV, Borisova TI, Gazaryan SV, Yager P, Novel lyssaviruses isolated from bats in Russia. Emerg Infect Dis. 2003;9:1623–5.PubMedGoogle Scholar
- Müller T, Johnson N, Freuling CM, Fooks AR, Selhorst T, Vos A. Epidemiology of bat rabies in Germany. Arch Virol. 2007;152:273–88. DOIPubMedGoogle Scholar
- Van der Poel WH, Van der Heide R, Verstraten ER, Takumi K, Lina PH, Kramps JA. European bat lyssaviruses, the Netherlands. Emerg Infect Dis. 2005;11:1854–9.PubMedGoogle Scholar
- Picard-Meyer E, Barrat J, Tissot E, Verdot A, Patron C, Barrat MJ, Bat rabies surveillance in France, from 1989 through May 2005. Dev Biol (Basel). 2006;125:283–8.PubMedGoogle Scholar
- Serra-Cobo J, Amengual B, Abellan C, Bourhy H. European bat lyssavirus infection in Spanish bat populations. Emerg Infect Dis. 2002;8:413–20. DOIPubMedGoogle Scholar
- Vázquez-Morón S, Juste J, Ibáñez C, Ruiz-Villamor E, Avellón A, Vera M, Endemic circulation of European bat lyssavirus type 1 in serotine bats, Spain. Emerg Infect Dis. 2008;14:1263–6. DOIPubMedGoogle Scholar
- Juste J, Bilgin R, Muñoz J, Ibáñez C. Mitochondrial DNA signatures at different spatial scales: from the effects of the Straits of Gibraltar to population structure in the meridional serotine bat (Eptesicus isabellinus). Heredity. 2009;103:178–87. DOIPubMedGoogle Scholar
- Ibáñez C, Garcia-Mudarra JL, Ruedi M, Stadelmann B. The Iberian contribution to cryptic diversity in European bats. Acta Chiropt. 2006;8:277–97. DOIGoogle Scholar
- Vázquez-Morón S, Avellón A, Echevarría JE. RT-PCR for detection of all seven genotypes of Lyssavirus genus. J Virol Methods. 2006;135:281–7. DOIPubMedGoogle Scholar
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.PubMedGoogle Scholar
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R, Dna SP. DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–7. DOIPubMedGoogle Scholar
- Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002;19:2092–100.PubMedGoogle Scholar
- Hughes GJ, Orciari LA, Rupprecht CE. Evolutionary timescale of rabies virus adaptation to North American bats inferred from the substitution rate of the nucleoprotein gene. J Gen Virol. 2005;86:1467–74. DOIPubMedGoogle Scholar
- Davis PL, Holmes EC, Larrous F, Van der Poel WH, Tjornehoj K, Alonso WJ, Phylogeography, population dynamics, and molecular evolution of European bat lyssaviruses. J Virol. 2005;79:10487–97. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 17, Number 3—March 2011
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors:
Top