Volume 17, Number 4—April 2011
Research
Molecular Epidemiology of Coxiella burnetii from Ruminants in Q Fever Outbreak, the Netherlands
Cite This Article
Citation for Media
Abstract
Q fever is a zoonosis caused by the bacterium Coxiella burnetii. One of the largest reported outbreaks of Q fever in humans occurred in the Netherlands starting in 2007; epidemiologic investigations identified small ruminants as the source. To determine the genetic background of C. burnetii in domestic ruminants responsible for the human Q fever outbreak, we genotyped 126 C. burnetii–positive samples from ruminants by using a 10-loci multilocus variable-number tandem-repeat analyses panel and compared them with internationally known genotypes. One unique genotype predominated in dairy goat herds and 1 sheep herd in the human Q fever outbreak area in the south of the Netherlands. On the basis of 4 loci, this genotype is similar to a human genotype from the Netherlands. This finding strengthens the probability that this genotype of C. burnetii is responsible for the human Q fever epidemic in the Netherlands.
Q fever is a zoonosis caused by Coxiella burnetii, an intracellular gram-negative bacterium that is prevalent throughout the world (1). Domestic ruminants are considered the main reservoir for Q fever in humans (2). However, other animal species, including pet animals, birds, and several species of arthropods, can be infected by C. burnetii and cause human cases of Q fever (2–5). The main clinical manifestations of Q fever in goats and sheep are abortion and stillbirth. In cattle, Q fever has been associated with sporadic abortion, subfertility, and metritis (4,6). With an abortion, up to 1 billion C. burnetii per gram of placenta can be excreted (7). Most animal species that carry C. burnetii show no symptoms (4). Transmission to humans occurs mainly through inhalation of contaminated aerosols (4,5,8–10).
Recently, 2 DNA-based methods for typing C. burnetii were reported (11–13). Multispacer sequence typing is based on DNA sequence variations in 10 short intergenic regions and can be performed on isolated C. burnetii strains or directly on extracted DNA from clinical samples (12,14,15). Multilocus variable-number tandem-repeat analyses (MLVA) is based on variation in repeat number in tandemly repeated DNA elements on multiple loci in the genome of C. burnetii and might be more discriminatory than multispacer sequence typing (13,15). MLVA also can be performed on C. burnetii strains (11,15) or directly on DNA extracted from clinical samples (16). A total of 17 different minisatellite and microsatellite repeat markers have been described (11).
Starting in 2007, the Netherlands has been confronted with one of the largest Q fever outbreaks in the world, involving 3,921 human cases in 4 successive years. On 28 dairy goat farms and 2 dairy sheep farms, abortion storms (with abortion rates up to 80%) caused by Q fever were diagnosed during 2005–2009. These small ruminants are considered the source of the human Q fever outbreak in the Netherlands (17). The connection between Q fever abortion storms in small ruminants and human Q fever cases is based primarily on epidemiologic investigations (18–21). A limited investigation by genotyping with MLVA recently showed that farms and humans in the Netherlands are infected by multiple different, yet closely related, genotypes of C. burnetii (16).
Although dairy goats and dairy sheep appear to be the source of the human Q fever outbreak in the Netherlands, no information is available about the genetic background of C. burnetii in these populations. This knowledge is essential for gaining insight into the molecular epidemiology of the organism and the origin of the outbreak, as well as for outbreak management purposes.
Our objective was to show the genetic background of C. burnetii in domestic ruminants responsible for the human Q fever outbreak. This information is necessary to evaluate the epidemiologic link between the source and human cases and to compare the outbreak genotypes with internationally known genotypes. During 2008–2010, a total of 125 C. burnetii–positive samples from 14 dairy goat farms, 1 dairy cattle farm, and 2 sheep farms were typed by MLVA. In addition, we show the geographic distribution of these C. burnetii genotypes across the Netherlands and compare the genotypes with what is internationally known.
Animal Samples
Figure 1
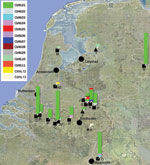
Figure 1. Map of the Netherlands showing locations of farms sampled during the Q fever outbreak, 2007–2010. Farms are indicated by letter and ruminant species (black squares, goats; black triangles, sheep; black star,...
Our study comprised 14 dairy goat farms (farms A–E, H, J, M, N, O, P, Q, AE, and AF), 1 dairy cattle farm (farm R), and 2 sheep farms (1 dairy sheep farm Y and 1 sheep farm Z) sampled during the Q fever outbreak in the Netherlands (Table 1; Figure 1). On 12 of the 14 dairy goat farms, multiple abortions had occurred. On 2 dairy goat farms (farms J and M) and on the dairy sheep farm (farm Y), no abortions had occurred. On 1 dairy cattle farm and on the sheep farm (farm Z), C. burnetii was detected in a placenta after abortion. One goat farm (farm AG) sampled in 2001 was included with an archived histologic section of paraffin-embedded placenta from an abortion outbreak caused by C. burnetii infection.
Vaginal swabs and milk samples from dairy goats and dairy sheep were sent to the national reference laboratory for notifiable animal diseases (the Central Veterinary Institute, part of Wageningen UR) by the Dutch Food and Consumer Product Safety Authority in accordance with the regulation in place at that time. These samples were submitted for confirmation testing of farms with clinically suspected Q fever (farms A–D, N, O, P, and Q), for tracing the source of human Q fever cases (because of proximity to human case-patients, farms H, J, and M) or for bulk tank milk monitoring (farm Y). Samples of immunohistochemically confirmed Q fever–positive goat and sheep placentas (farms N, AE, AF, and Z) and fetal tissue (farm E) were provided by the Animal Health Service, including 1 archived histologic section of paraffin-embedded placenta from a C. burnetii abortion outbreak in a goat farm in 2001 (farm AG), which was diagnosed retrospectively (22). The sampled dairy goat farms represent 60% of the farms with known abortion problems during 2007–2009.
Testing of Samples before MLVA Typing
DNA was extracted from vaginal swabs and milk by using Chelex resin (InstaGene; Bio-Rad, Hercules, CA, USA). A vaginal swab tip or 200 μL of milk was added to 400 μL of Chelex suspension and incubated and shaken for 30 min at 56°C, followed by an incubation step for 8 min at 100°C. The clarified supernatant was used for PCR and MLVA. DNA from placentas was extracted by using a DNA tissue kit (DNeasy Blood and Tissue Kit; QIAGEN, Hilden, Germany). DNA from the paraffin-embedded placenta was extracted by using MagneSil Genomic Fixed Tissue System (Promega, Madison, WI, USA).
All samples were tested by an in-house real-time PCR directed toward the C. burnetii–specific IS1111a element (23). An inhibition control was constructed by using the primes of the IS1111a element (Table 2). PCR was performed on a 7500 Fast Real Time PCR system (Applied Biosystems, Foster City, CA, USA) by using 400 nmol/L of primers and 200 nmol/L of probes in 7 μL PerfeCTa Multiplex qPCR Supermix, uracil-
MLVA Typing
MLVA typing was performed by using a selection of 10 of the 17 loci described by Arricau-Bouvery et al. (11) according to the Multiple Loci VNTR Analysis databases for genotyping (http://minisatellites.u-psud.fr/MLVAnet/querypub1.php), except that Ms12 was omitted because of poor performance, and Ms24 was added (Table 2). New primers were designed for Ms27 and Ms28 to improve performance. The annotation of Ms30, Ms31, and Ms36 was updated (P. Le Flèche, pers. comm.).
The PCR amplification was performed by using an Applied Biosystems 9700 thermocycler in a total volume of 25 µL containing 1× reaction buffer, 1 U True Start Taq DNA polymerase (Fermentas, Glen Burnie, MD, USA), 2 mmol/L MgCl2, 0.2 mmol/L of each nucleotide (dATP, dGTP, dCTP, dUTP), 0.5 µmol/L of each primer, 0.5 U UDG (New England Biolabs), and 2–5 µL template. An initial UDG incubation for 5 min at 37°C and denaturation/activation for 2 min at 95°C was followed by 40 cycles of denaturation for 30 s at 95°C, annealing for 30 s at 60/65°C, elongation for 30 s at 72°C, followed by a final extension step for 5 min at 72°C. After the amplification, 0.5 U UDG inhibitor (New England Biolabs) was added to the PCRs to prevent further UDG activity. Up to 4 different PCR products with different fluorescent dyes were diluted, depending on the PCR efficiency, and pooled. From these pooled PCR products, 4 µL was mixed with 15 µL of Hi-Di formamide (Applied Biosystems) and 0.5 µL of GeneScan 600 LIZ Size Standard (Applied Biosystems). After denaturation for 3 min at 96°C the samples were cooled on ice. The PCR products were separated on a 3130 Genetic Analyzer (Applied Biosystems) with a 36-cm array by using POP7 polymer.
The fragments were sized by using GeneMapper version 4.0 software (Applied Biosystems). The accuracy of the sizing obtained by capillary electrophoresis was determined by comparing sequencing data from the reference strain with the obtained fragment size from the capillary electrophoresis and corrected if necessary. The number of repeats for each locus was determined on the basis of the published and corrected annotation of the various loci (Table 2). Non–whole repeat numbers were rounded off mathematically. Reproducibility was checked with positive controls.
Data Analysis
The reference strain Nine Mile was used as reference (11). Analyses were performed, including only genotypes of C. burnetii containing <2 loci with missing values. Numerical typing data were imported into BioNumerics v 6.1 (Applied Maths, Sint-Martens-Latem, Belgium) and analyzed with the multistate categorical similarity coefficient by using unweighted pair group method with arithmetic mean clustering. Missing values were imported as question marks. The genotypic diversity of the population under study was calculated by using the adapted Simpson index of diversity (Hunter-Gaston diversity index [HGDI]) (11,24).
Found MLVA patterns based on the number of repeats per locus were called MLVA types and identified as CbNLxx. We compared MLVA types with MLVA types in the publicly accessible Multiple Loci VNTR Analysis databases for genotyping: Coxiella2007 and Coxiella2009_Netherlands (access date 2001 Jan 11). The Nine Mile strain was used as reference.
The study comprised 122 samples from 15 dairy goat farms, 2 samples from 1 dairy sheep farm, and 1 sample each from 1 sheep farm and 1 dairy cattle farm were included in this study (Table 1). Of the farms sampled during the outbreak, 13 were situated in the southern part of the Netherlands; 3 dairy goat farms (farms M, N, and AE) and 1 dairy sheep farm (farm Y) were located outside this area (Figure 1). From the 238 Q fever PCR-positive samples from the farms in this study, 125 (53%) yielded a genotype with <2 missing values: 52 with a complete genotype, 48 with 1 missing value, and 25 with 2 missing values. 113 (47%) PCR-positive samples represented partial genotypes with 3–10 missing values. From the paraffin-embedded placenta (farm AG), only a partial genotype could be shown, with 6 repeats on Ms03 and 10 repeats on Ms34.
Figure 2

Figure 2. Phylogenetic tree with genotypes of Coxiella burnetii of all samples in the study, the Netherlands, on the basis of 10 multilocus variable-number tandem-repeat analyses (MLVA). Repeats per locus are shown; open...
We distinguished 13 genotypes in the 125 samples (CbNL01–CbNL13; Table 1; Figures 1, 2). All C. burnetii genotypes could be associated with abortion, except for 2 (CbNL10, farm M and Y; and CbNL08, farm J; Figure 1). The relationship between the genotypes in all samples is shown in Figure 2, including the genotype of the reference strain Nine Mile and the reference genotype of the reference strain Nine Mile from Arricau-Bouvery et al. (11), which were identical.
Figure 3
![Thumbnail of Phylogenetic tree with genotypes of Coxiella burnetii that are most closely related to the Dutch genotypes on the basis of 4 multilocus variable-number tandem-repeat analyses (MLVA). Genotypes are derived from the Multiple Loci VNTR Analysis databases for genotyping (http://minisatellites.u-psud.fr/MLVAnet/querypub1.php: Coxiella2009_Netherlands [accessed 2011 Jan 11]). Repeats per locus are shown; open spots indicate missing values. NL, the Netherlands.](/eid/images/10-1562-F3-tn.jpg)
Figure 3. Phylogenetic tree with genotypes of Coxiella burnetii that are most closely related to the Dutch genotypes on the basis of 4 multilocus variable-number tandem-repeat analyses (MLVA). Genotypes are derived from the...
Figure 4
![Thumbnail of Phylogenetic tree with genotypes of Coxiella burnetii that are most closely related to the Dutch genotypes on the basis of 9 multilocus variable-number tandem-repeat analyses (MLVA). Genotypes are derived from the Multiple Loci VNTR Analysis databases for genotyping (http://minisatellites.u-psud.fr/MLVAnet/querypub1.php: Coxiella2007 [accessed 2011 Jan 11]). Repeats per locus are shown; open spots indicate missing values. NL, the Netherlands; Slovak Rep, Slovak Republic.](/eid/images/10-1562-F4-tn.jpg)
Figure 4. Phylogenetic tree with genotypes of Coxiella burnetii that are most closely related to the Dutch genotypes on the basis of 9 multilocus variable-number tandem-repeat analyses (MLVA). Genotypes are derived from the...
The 13 genotypes are separated in 2 clusters (Figure 2). One cluster containing a genotype represented by 111 (90%) of the samples (CbNL01); 1 genotype (CbNL10) represented by 3 samples (1 from a dairy goat farm and 2 from a dairy sheep farm); and 10 genotypes (CbNL02–CbNL09 and CbNL11) represented by 1 sample, all from dairy goat farms. The second cluster was distinctly separated from the other cluster, representing 2 genotypes in 1 dairy goat sample (CbNL12), in 1 dairy cattle sample (CbNL13) and the paraffin-embedded placenta. In samples from dairy goat farms with abortion problems, the same genotype (CbNL01) was present in 110 (91%) of 121 samples. One sheep sample also showed this genotype (farm Z). The geographic distribution of the genotypes according to the location of the originating farm is given in Figure 1. The relationship between the genotypes found in this study and the internationally known genotypes are presented in the phylogenetic trees in Figure 3 on the basis of 4 loci and in Figure 4 on the basis of 9 loci.
We performed MLVA typing of C. burnetii based on 10 loci on a large number of Q fever–positive samples to show the genetic background of C. burnetii in the domestic ruminants associated with the Q fever outbreak in humans in the Netherlands. In 125 (53%) of 237 samples, an adequate genotype for C. burnetii was generated. Previously, MLVA typing was performed on C. burnetii strains after primary isolation and cultivation (11,13,15) or, in the Netherlands, on only 11 clinical samples from humans, sheep, and goats with a selected number of 3 loci (16).
The main drawback of typing on clinical samples is the variable quality and amount of DNA. These drawbacks influence the typability of samples, resulting in partial genotypes; whether the missing values are caused by insufficient DNA concentrations and quality or by an absence of loci is unclear. If loci are absent, partial genotypes also are expected to be found in samples with high DNA loads. Such is not the case in our study. Typing of placenta material that contains high quantities of C. burnetii, as well as vaginal swabs with PCR cycle threshold (Ct) values <32, yielded complete genotypes. In samples with Ct values of 32–34, only partial genotypes were obtained. Samples with a Ct value >34 were poorly typable.
Arricau-Bouvery et al. (11) calculated diversity indices for the 17 loci used in the MLVA, which varied from 0.28 for locus Ms22 to 0.86 for locus Ms34. The HGDI for the combined panels 1 and 2 of the MLVA typing method for C. burnetii can be calculated on 0.99 and for panel 2 on 0.92. These HGDIs are in the upper part of the 0.438–0.997 range reported by Hunter and Gaston (24) for typing methods for various bacteria and yeasts.
The high diversity indices for the MLVA of C. burnetii indicate a high discriminating power, and this capability makes MLVA typing suitable for distinguishing C. burnetii isolates. With this highly discriminatory typing method, we found that 1 genotype of C. burnetii predominated on all dairy goat farms in the southern part of the Netherlands. On 12 of 14 dairy goat farms, this genotype was found in 91% of samples, varying per farm from 33% (farm E) to 100% (farms B, C, N–Q, Table 1, Figures 1 and 2). Although the sample size was small compared with the number of animals on the farm (Table 1), these data show that 1 genotype was far more common than other genotypes found on these farms. The 9 other genotypes occurred once, each representing only 0.8% of all found genotypes on dairy goat farms. The most predominant genotype was found on all 11 dairy goat farms in the southern Netherlands and on a farm in the eastern part of the country (farm N). This finding strongly suggests a clonal spread of C. burnetii with this predominant genotype over the dairy goat farms in the southeastern part of the Netherlands.
The clonal spread of 1 genotype of C. burnetii could be explained by 2 phenomena. First, the dairy goat industry in the Netherlands sharply increased from almost 100,000 dairy goats in 2000 to >230,000 dairy goats on ≈350 farms in 2009 (17). Most of these goats were bred in the Netherlands, which probably resulted in a microbial relationship between many of the dairy goat herds. In this theory, the C. burnetii strain with the most predominant genotype was present in the Netherlands for a long period before the abortion problems in dairy goats started in 2005. This theory is not supported by the results of the typing of the paraffin-embedded placenta from an aborted dairy goat who in 2001. The typing result differs on 2 loci from the most predominant genotype found in this study. Second, clonal spread could have been facilitated by emergence of a genotype of C. burnetii causing abortion in dairy goats that could then spread successfully over the dense goat population in the southeastern part of the country. Whether this genotype is more virulent is subject to research.
On the basis of comparison of MLVA types on 4 loci (Figure 3), CbNL01–06 could not be distinguished and were similar to the genotype of a person in the Netherlands (QPK2) and 2 genotypes from persons in France (Cb#88, Cb#97). The sample from a person in the Netherlands is derived from patient 2 reported by Klaassen et al. (16). Patient 2 is the farmer of farm A, where genotype CbNL01 predominated, as well as CbNL05 (Table 1). This shows a genetic link between the C. burnetii DNA from the farmer and his abortive goats, which suggests that the farmer was infected by his own goats. However, this link is based on only 4 loci on 1 human sample. To further confirm the link between dairy goats and humans, more samples need to be typed with more MLVA loci to increase the discriminatory power.
The human sample with ID QKP6 is the same sample as that from patient 4 reported by Klaassen et al. (16) and is most closely related to CbNL07. Human sample QKP1 is the same as that of patient 1. Patient 5 fits in the genotype cluster in the Netherlands, as does patient 2. The sheep reported by Klaassen et al. did not abort, and their samples show a difference of 1 repeat on Ms34 compared with CbNL01. On the basis of the comparison of MLVA types on 9 loci (Figure 4), all genotypes in this study can be distinguished. The most predominant genotype CbNL01 clusters with other genotypes (CbNL02–CbNL09, CbNL11) and with 1 human sample (Cb#97) from France. CbNL01 differed from this human isolate on 2 loci (Ms30 and 36), which shows that the most predominant genotype in the Netherlands is unique. Whether this finding can be attributed to the small number of strains and clinical samples typed or is really a unique genotype is not yet clear. The closest relation to an isolate from France might give a clue about the origin of the genotype from the Netherlands.
The human Q fever outbreak in the Netherlands started in the southern part of the country and resulted in >3,500 human cases during 2007–2010. Dairy goats and dairy sheep are considered to be the source of this outbreak, primarily on the basis of epidemiologic findings (10,17,20,21,25,26). In our study, samples were typed from farms suspected of being the source of the human Q fever outbreak. Results show that 1 genotype of C. burnetii predominated in the dairy goats and sheep in the human Q fever outbreak area in the southern part of the Netherlands, and this genotype also was present in a human case-patient in the Netherlands. This C. burnetii genotype is expected to have played a key role in the Q fever outbreak in small ruminants in the Netherlands and was also transmitted widely to humans, causing Q fever in the human population. If this hypothesis holds true, C. burnetii with the same genotype as in dairy goats should be found in most samples from human Q fever patients. To this end, a study was performed to show the genetic background of human C. burnetii isolates in the Netherlands by using a concordant MLVA typing method (J.J.H.C. Tilburg et al., unpub. data). Furthermore, the uniqueness of the predominant genotype of C. burnetii for the Netherlands can be part of the explanation why the magnitude of the Q fever outbreak in the Netherlands has never been seen elsewhere.
Dr Roest is a veterinarian at the Central Veterinary Institute, part of Wageningen UR. He is project leader for the Q fever in goats project, was active in the management of the Q fever outbreak in the Netherlands, and has a special interest in zoonotic bacterial diseases.
Acknowledgments
We thank Gert Jan Boender and Fred van Welie for preparing the graphs. We also thank N. Groot Niebelink for providing the Q fever PCR-positive placenta from an abortive cow (farm R) and D. Frangoulidis for providing the C. burnetii reference strain Nine Mile RSA 493.
This study was supported by the Dutch Ministry of Agriculture, Nature and Food Quality.
References
- Raoult D, Marrie TJ, Mege JL. Natural history and pathophysiology of Q fever. Lancet Infect Dis. 2005;5:219–26. DOIPubMedGoogle Scholar
- Woldehiwet Z. Q fever (coxiellosis): epidemiology and pathogenesis. Res Vet Sci. 2004;77:93–100. DOIPubMedGoogle Scholar
- Babudieri B, Moscovici C. Experimental and natural infection of birds by Coxiella burneti. Nature. 1952;169:195–6. DOIPubMedGoogle Scholar
- Arricau-Bouvery N, Rodolakis A. Is Q fever an emerging or re-emerging zoonosis? Vet Res. 2005;36:327–49. DOIPubMedGoogle Scholar
- Muskens J, Mars MH, Franken P. Q fever: an overview [in Dutch]. Tijdschr Diergeneeskd. 2007;132:912–7.PubMedGoogle Scholar
- Arricau Bouvery N, Souriau A, Lechopier P, Rodolakis A. Experimental Coxiella burnetii infection in pregnant goats: excretion routes. Vet Res. 2003;34:423–33. DOIPubMedGoogle Scholar
- Marrie TJ. Q fever. Volume I: the disease. Boca Raton (FL): CRC Press; 1990.
- Schimmer B, Dijkstra F, Vellema P, Schneeberger PM, Hackert V, ter Schegget R, Sustained intensive transmission of Q fever in the south of the Netherlands, 2009. Euro Surveill. 2009;14: pii: 19210.
- Arricau-Bouvery N, Hauck Y, Bejaoui A, Frangoulidis D, Bodier CC, Souriau A, Molecular characterization of Coxiella burnetii isolates by infrequent restriction site-PCR and MLVA typing. BMC Microbiol. 2006;6:38. DOIPubMedGoogle Scholar
- Glazunova O, Roux V, Freylikman O, Sekeyova Z, Fournous G, Tyczka J, Coxiella burnetii genotyping. Emerg Infect Dis. 2005;11:1211–7.PubMedGoogle Scholar
- Svraka S, Toman R, Skultety L, Slaba K, Homan WL. Establishment of a genotyping scheme for Coxiella burnetii. FEMS Microbiol Lett. 2006;254:268–74. DOIPubMedGoogle Scholar
- Mediannikov O, Fenollar F, Socolovschi C, Diatta G, Bassene H, Molez JF, Coxiella burnetii in humans and ticks in rural Senegal. PLoS Negl Trop Dis. 2010;4:e654. DOIPubMedGoogle Scholar
- Chmielewski T, Sidi-Boumedine K, Duquesne V, Podsiadly E, Thiery R, Tylewska-Wierzbanowska S. Molecular epidemiology of Q fever in Poland. Pol J Microbiol. 2009;58:9–13.PubMedGoogle Scholar
- Klaassen CH, Nabuurs-Franssen MH, Tilburg JJ, Hamans MA, Horrevorts AM. Multigenotype Q fever outbreak, the Netherlands. Emerg Infect Dis. 2009;15:613–4. DOIPubMedGoogle Scholar
- Roest HI, Tilburg JJ, van der Hoek W, Vellema P, van Zijderveld FG, Klaassen CH, The Q fever epidemic in the Netherlands: history, onset, response and reflection. Epidemiol Infect. 2011;139:1–12. DOIPubMedGoogle Scholar
- Van Steenbergen JE, Morroy G, Groot CA, Ruikes FG, Marcelis JH, Speelman P. An outbreak of Q fever in the Netherlands–possible link to goats [in Dutch]. Ned Tijdschr Geneeskd. 2007;151:1998–2003.PubMedGoogle Scholar
- Koene RP, Schimmer B, Rensen H, Biesheuvel M, de Bruin A, Lohuis A, A Q fever outbreak in a psychiatric care institution in the Netherlands. Epidemiol Infect. 2011;139:13–8. DOIPubMedGoogle Scholar
- Karagiannis I, Schimmer B, Van Lier A, Timen A, Schneeberger P, Van Rotterdam B, Investigation of a Q fever outbreak in a rural area of the Netherlands. Epidemiol Infect. 2009;137:1283–94. DOIPubMedGoogle Scholar
- Karagiannis I, Morroy G, Rietveld A, Horrevorts AM, Hamans M, Francken P, Q fever outbreak in the Netherlands: a preliminary report. Euro Surveill. 2007;12:E070809.2. PMID: 17868551
- Wouda W, Dercksen DP. Abortion and stillbirth among dairy goats as a consequence of Coxiella burnetii [in Dutch]. Tijdschr Diergeneeskd. 2007;132:908–11.PubMedGoogle Scholar
- Klee SR, Tyczka J, Ellerbrok H, Franz T, Linke S, Baljer G, Highly sensitive real-time PCR for specific detection and quantification of Coxiella burnetii. BMC Microbiol. 2006;6:2. DOIPubMedGoogle Scholar
- Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol. 1988;26:2465–6.PubMedGoogle Scholar
- Schimmer B, Morroy G, Dijkstra F, Schneeberger PM, Weers-Pothoff G, Timen A, Large ongoing Q fever outbreak in the south of the Netherlands, 2008. Euro Surveill. 2008;13:pii:18939.
- van der Hoek W, Dijkstra F, Schimmer B, Schneeberger PM, Vellema P, Wijkmans C, Q fever in the Netherlands: an update on the epidemiology and control measures. Euro Surveill. 2010;15: pii: 19520.
Figures
Tables
Cite This ArticleTable of Contents – Volume 17, Number 4—April 2011
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Hendrik I.J. Roest, Central Veterinary Institute of Wageningen UR, Edelhertweg 15, 8219 PH Lelystad, the Netherlands
Top