Volume 19, Number 4—April 2013
Letter
Hepatitis E Virus Genotype 3 Strains in Domestic Pigs, Cameroon
Figure
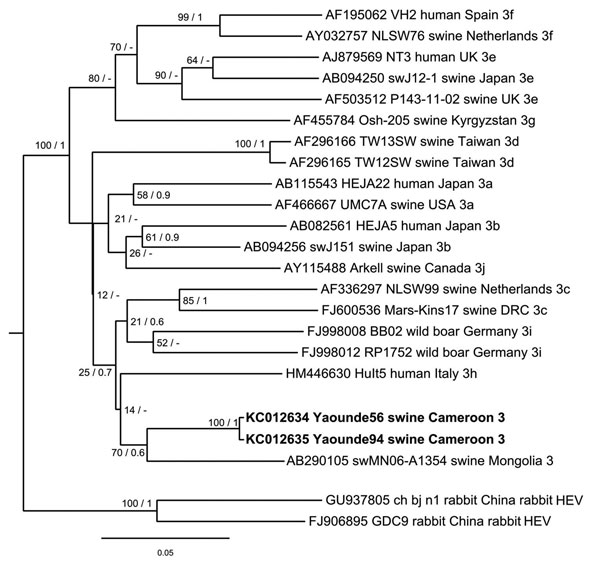
Figure. . . . . . Phylogenetic analysis of hepatitis E virus (HEV) strains, Cameroon. The Bayesian phylogenetic tree was constructed by using partial nucleotide sequence of open reading frame 2 (278 nt) of HEV. For each sequence used, the GenBank accession number, strain designation, source of isolation, country of isolation, and HEV subtype are shown. Multiple nucleotide sequence alignment was analyzed by using the Markov Chain Monte Carlo method implemented in the program MrBayes version 3.0 (http://mrbayes.sourceforge.net/) and applying the general time-reversible substitution model. Posterior probabilities are shown at the nodes of the tree to the right of the slash if >0.5. Bootstrap values calculated from 10,000 replicates are indicated at the nodes of the tree to the left of the slash. Alignment was analyzed by using the neighbor-joining method and resulted in same tree topology (not shown). Newly described HEV sequences are shown in boldface. Scale bar indicates evolutionary distance. UK, United Kingdom; USA, United States; DRC, Democratic Republic of Congo.
1These authors contributed equally to this article.