Volume 19, Number 5—May 2013
Research
Full-Genome Deep Sequencing and Phylogenetic Analysis of Novel Human Betacoronavirus
Matthew Cotten, Tommy T. Lam, Simon J. Watson, Anne L. Palser, Velislava Petrova, Paul Grant, Oliver G. Pybus, Andrew Rambaut, Yi Guan, Deenan Pillay, Paul Kellam , and Eleni Nastouli
, and Eleni Nastouli
Figure 3
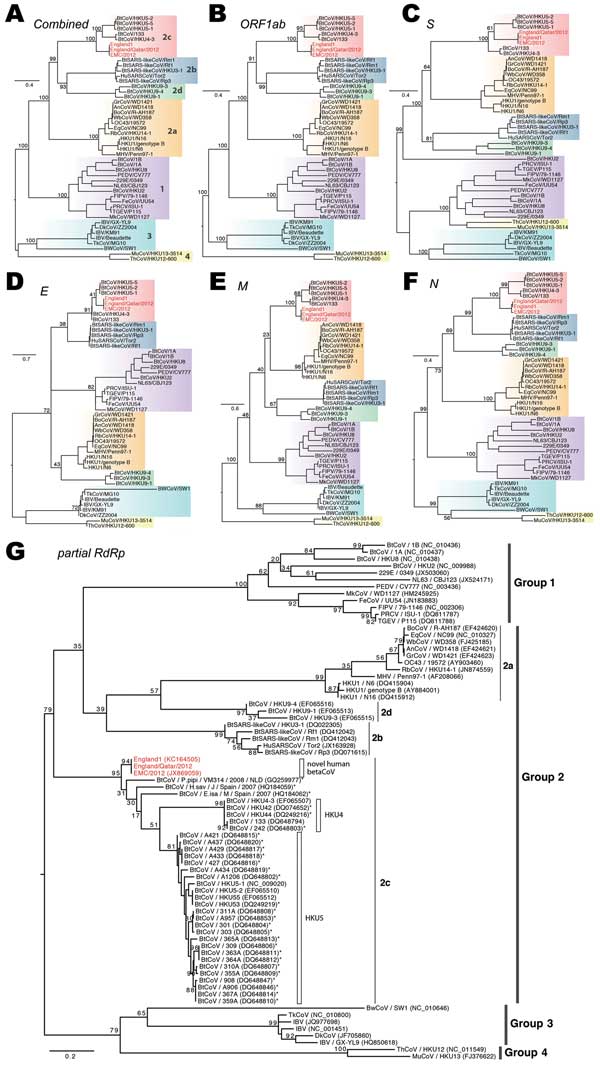
Figure 3. . . . Phylogenetic analyses of coronaviruses. A–F) Maximum-likelihood phylogenies of combined and each individual open reading frame (ORF), including ORF 1ab, S, E, M, and N. Previously defined viral lineages (group 1, 2a, 2b, 2c, 2d, 3, and 4) are highlighted by color blocks and described in (A). G) Phylogenetic analyses on the partial RNA-dependent RNA polymerase sequence region (396 bp) of coronaviruses (CoVs). Partial gene sequences from other CoVs that are closely related to the novel human betaCoVs are included and marked with asterisks. Bootstrap analysis of 1,000 replicates was performed for each phylogeny. The novel human betaCoVs studied here are shown in red. Scale bar indicates nucleotide substitutions per site.
Page created: April 23, 2013
Page updated: April 23, 2013
Page reviewed: April 23, 2013
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.